











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Antecedentes
Las inmunodeficiencias primarias o errores innatos de la inmunidad (EII) son enfermedades genéticas que producen defectos cuantitativos o funcionales de diferentes moléculas del sistema inmunológico.1
Los EII pueden presentarse de diferente forma y deben sospecharse en los pacientes con antecedente familiar de EII, enfermedades graves o de presentación temprana en la vida, infecciones recurrentes/graves (por ejemplo neumonías complicadas, bronquiectasias, sinusitis y otitis supurativa), uso de antibióticos parenterales, gérmenes oportunistas o atípicos, diarrea crónica, falla de medro, abscesos profundos o de piel, complicaciones por aplicación de vacunas vivas atenuadas (BCG), enfermedades autoinmunes o alérgicas de curso clínico insidioso, mala respuesta al tratamiento, así como tendencia a trastornos oncológicos.1
La tasa global de prevalencia de EII en Europa es de 1:16 000 a 1:50 000 habitantes.2 La incidencia en Estados Unidos y Argentina es de 1:10 000 a 1:20 000 nacidos vivos mientras que en Brasil es de 1:146 000 y en México de 1:98 0000 nacidos vivos.3
Actualmente se han descrito más de 430 defectos monogénicos; en 2019 fueron clasificados en el 2019 en 10 grupos por un comité de expertos de la IUIS (International Union of Immunological Societies) a continuación se enlistan.1,4
Inmunodeficiencias que afectan la inmunidad celular y humoral.
Inmunodeficiencias combinadas asociadas con hallazgos sindromáticos.
Deficiencias predominantemente de anticuerpos.
Enfermedades de desregulación inmune.
Defectos en fagocitos en número, función o en ambos.
Defectos en la inmunidad intrínseca e innata.
Desórdenes autoinflamatorios.
Deficiencias del complemento.
Falla en la médula ósea.
Fenocopias de inmunodeficiencias primarias.
En cuanto a la frecuencia de los EII, las deficiencias predominantemente de anticuerpos siguen siendo el grupo más frecuente (51.9 %), seguidos de los síndromes bien definidos (14.5 %), inmunodeficiencia combinadas (9.9 %), defectos de fagocitosis (7.9 %), entre otros.5
El registrar los casos de EII es una herramienta útil para obtener parámetros epidemiológicos. Los datos objetivos como la incidencia y prevalencia de los EII en una población ayudan a establecer nuevos programas de salud dirigidos a mejorar el diagnóstico y tratamiento de estas enfermedades. La European Society for Immunodeficiencies (ESID) se formó como un grupo informal en 1983 y en 1994 se estableció como la sociedad que actualmente es. Entre sus objetivos están el registrar a cada uno de los pacientes con EII que se tratan en los hospitales en Europa. Hasta agosto del 2020, el grupo ESID reporta 33 221 casos de EII. La frecuencia de la distribución de los EII del registro de ESID es el siguiente: deficiencias predominantemente de anticuerpos 51.8 %, síndromes bien definidos 14.5 %, inmunodeficiencias combinadas 9.9 % y defectos de la fagocitosis 7.9 %.5
En el año 2009 se consolidó la Latin American Society for Immunodeficiencies, (LASID), que surgió inicialmente como Latin American Group for Primary Immunodeficiency Disease en 1993 y emitió su primer reporte de registro de casos de EII en 1998, participaron Argentina, Brasil, Colombia, Costa Rica, Chile, México, Paraguay y Uruguay con un total de 1428 casos. En ese reporte los pacientes mexicanos que se registraron fueron procedentes de un solo hospital, el Instituto Nacional de Pediatría (INP) con 118 pacientes.6 En febrero del 2015 LASID creó una plataforma de registro en la cual todos los países de Latinoamérica que así lo decidan pueden participar registrando los casos de EII. En cada país, cada hospital puede ser un centro que registre casos de EII, de modo que no hay un límite en el número de centros hospitalarios afiliados a LASID por país.7
Para que un hospital pueda afiliarse como un centro de registro de EII en la plataforma de LASID, debe solicitar su afiliación enviando un correo electrónico a lasidregistry@gmail.com. a cada centro afiliado se le asigna un número de usuario y clave de acceso. Cada paciente que se registra en línea en la plataforma de LASID, se cataloga por grupo de EII y se completan algunos datos demográficos, clínicos y de laboratorio. LASID contabiliza mensualmente el número de EII registrados por país y centro, de modo que podemos conocer la información actualizada mensualmente de Latinoamérica, de cada país y de nuestro hospital desde el cual registramos.
Actualmente son 18 los países que registran EII en el registro de LASID: Argentina, Bolivia, Brasil, Chile, Colombia, Costa Rica, Cuba, República Dominicana, Ecuador, El Salvador, Guatemala, Honduras, México, Panamá, Paraguay, Perú, Uruguay y Venezuela. Hasta hoy se han registrado 8,329 casos de EII en Latinoamérica. En México 46 hospitales distribuidos en todo el país son centros de registro de EII afiliados a LASID. En México hasta julio de 2020 se han registrado 1,815 pacientes (Figura 1).7 Este estudio describe el registro de EII en un hospital de México y hace una revisión del registro de EII a nivel internacional y nacional.

Registro de EII en un hospital pediátrico de Sinaloa
El Hospital Pediátrico de Sinaloa (HPS) está ubicado en una ciudad en la región Noroeste de México, es un nosocomio que tiene las principales especialidades médicas y quirúrgicas de pediatría, cuenta con 92 camas, formas especialistas en pediatría y registra alrededor de 19 348 consultas por año.8
Desde marzo del 2017 el HPS integró la especialidad de inmunología clínica, y con ello se inició el diagnóstico de los EII. Después del diagnóstico del primer EII el HPS se integró como un centro de registro de EII afiliado a LASID. En tres años hemos diagnosticado 12 casos de EII, en la Figura 2 se describen según la clasificación de la IUIS, las deficiencias predominantemente de anticuerpos fueron las más frecuentes.
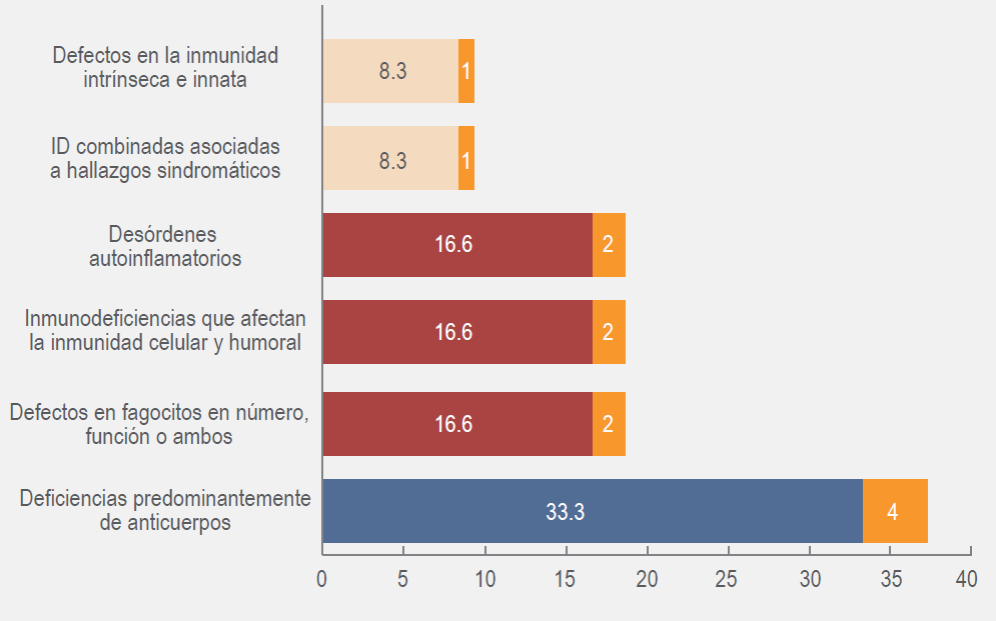
Figura 2. Distribución del tipo de los errores innatos de la inmunidad del Hospital Pediátrico de Sinaloa.
En el Cuadro 1 se describen de forma específica las características de la presentación clínica, hallazgos de laboratorio y radiológicos que orientaron el diagnóstico, basados en los criterios diagnósticos bien establecidos de EII por ESID (https://esid.org/Education/Diagnostic-Criteria-PID).9, 10 Llama la atención que en ningún paciente se encontraron antecedentes familiares de EII, endogamia, ni consanguinidad, los cuales son datos característicos que hacen sospechar en un EII. El rango de edad al momento del diagnóstico fue de los 2 meses a 16 años, con un rango edad al inicio de los síntomas fue entre 19 días y 4 años. El sexo masculino predominó en 66.6 %, con una relación M:F de 1.33. El 90 % de los casos se presentaron con infecciones y 8.3 % con eventos autoinmunes. El 83.3 % de los casos recibieron inmunoglobulina intravenosa (IgIV), principalmente aquellos con defectos predominantes de anticuerpos e inmunodeficiencias combinadas, mientras que las inmunodeficiencias asociadas con síndromes y defectos de fagocitosis recibieron IgIV a dosis de inmunomodulación. El trasplante de células progenitoras hematopoyéticas (TCPH) se realizó el paciente con inmunodeficiencia combinada grave (IDCG). El diagnóstico genético en un caso se realizó en el Laboratorio de Inmunología Molecular de la Facultad de Medicina de Morelos, México. En el paciente con síndrome de Wiskott-Aldrich, se halló una variante patogénica en el gen WAS p.(Val345Tyrfs*100), ya fue reportado previamente.11 El 33.3 % de los 12 casos tuvo desenlace fatal; el paciente con IDCG con TCPH tuvo falla del injerto y falleció por choque séptico de foco abdominal; el paciente con síndrome de Wiskott-Aldrich falleció por ruptura de aneurisma y choque hipovolémico. Dos pacientes con defecto de fagocitosis desarrollaron síndrome de activación macrofágica refractario al tratamiento.
Cuadro 1. Características clínicas, hallazgos de laboratorio y gabinete relevantes, evolución, tratamiento y pronóstico de los pacientes con EII en el Hospital Pediátrico de Sinaloa
| IDP | Edad | Sexo | EIS | Edad | Alteraciones inmunológicas | Infecciones y germen aislado | Complicaciones | Tratamiento | Pronóstico |
| Inmunodeficiencias que afectan la inmunidad celular y humoral | 8 m | M | 19 d | 2 m | Linfopenia 830 células/ μL (2500- 4000 célulasl/μL), IgG 100 mg/dL (215- 601 mg/dL). TREC con sospecha de IDCG, Inmunofenotipo T- B- NK+ | IVU (E. coli), Candidiasis oral. Norovirus posTCPH | TCPH fallido. Sepsis abdominal con perforación intestinal | IgIV, TMPSMX, fluconazol, TCPH (4 meses) | Muerte a los 8 meses |
| 5 a | F | 8 m | 5 a | Linfocitos CD8+ 47 células/μL (330- 2200 células/μL). Relación CD4+/CD8+ 8.53 (1.0-3.2) | Rotavirus Neumonía nosocomial, IVU (P. aeruginosa) |
Bronquiectasias. Acropaquias. Desnutrición |
IgIV, TMPSMX, fluconazol | Vive | |
| Inmunodeficiencias combinadas asociadas con hallazgos sindromáticos | 7 a* | M | 3 m | 5 a | Plaquetas 15 000 células/ μL (150- 450 000 células/μL), volumen
plaquetario medio 7.5 fL (7.5-9), Coombs directo 1:8, IgG 352
mg/dL (463- 1236 mg/dL), IgA 80 mg/dL (25-154 mg/ dL), IgM 67
mg/dL (43-196 mg/dL), IgE 67.06 (0-60 UI/L). Linfocitos CD4+ 317 células/μL (560-2700 cél/μL), CD19+ 56 células/μL (220-1300 cél/μL). Variante patogénica en el gen de WAS p.(Val345Tyrfs*100) |
OMA repetición (3). Neumonía. Varicela complicada. Celulitis en pierna izquierda |
Uveítis Infarto en núcleo caudadoputamen izquierdo. Aneurisma aórtico gigante |
IgIV, TMPSMX, fluconazol, CsA, prednisona, | Muerte por hemorragia masiva a los 7 años |
| Deficiencias predominantemente de anticuerpos | 18 a | F | 4 a | 16a | IgG 137 mg/dL (639-1349 mg/dL), CD19+: 0.2 % (6- 23 %), CD16+/56+: 12 % (3-7 %), CD4+ CD45RO+ elevados | OMA supurativa (2) Sinusitis maxilar (3). Pansinusitis |
EVC hemorrágico. Colelitiasis. Hipoacusia bilateral |
IgIV, TMP-SMX, fluconazol Topiramato | Vive |
| Defectos de fagocitos en número, función o ambos | 15 a | M | 4 a | 12a | IgG 136 mg/dL (639-1349 mg/dL), CD19+/20+ : 4 % (6-23 %), CD4+ CD45RA+: 8 % (41/mm3), CD4+ CD45RO+: 67 % (346/mm3), CD21low: 21 % (22/mm3), células B CD27+ ausentes | GEPI recurrente (5). Neumonía (4) con derrame pleural. OMA (3). Sinusitis maxilar |
Bronquiectasias. Acropaquias, neumopatía crónica. Desnutrición moderada |
IgIV, TMP-SMX, fluconazol, CSI, oxígeno 0.5 L/ minuto | Vive |
| 7 a | M | 4 a | 5 a | Deficiencia en la respuesta en 78 % para PCV23 | Sinusitis maxilar (4). Neumonía con empiema y derrame pleural |
Bronquiectasias | IgSC por 4 meses | Vive | |
| 4 a | F | 12 m | 2 a | Deficiencia en la respuesta en 60 % para PCV23 | Sinusitis maxilar (4). OMA supurativa (2). Neumonía bacteriana (6) |
Asma. Rinitis alérgica. Desnutrición leve |
IgIV mensual por 6 meses. CSI + LABA/ montelukast / ITE | Vive | |
| 2 a | M | 2 a | 2 a | Hb 7.7 g/dL (11.5-14 g/dL), leucocitos 2000 cél/μL (4000- 10 000 cél/μL), neutrófilos 1080 cél/μL, (1500-6000 cél/μL), linfocitos 7601 500 cél/μL (1500-6000 cél/μL), plaquetas 67 000cél/ μL (150-450 000 cél/ μL), fibrinógeno 136 mg/dL (> 150 mg/dL) ferritina 9863 μg/L (300 μg/L), MO con hemofagocitosis | Neumonía. Sepsis. Abscesos hepáticos múltiples |
Síndrome de activación macrofágica. CID. Choque séptico |
Meropenem, vancomicina, fluconazol, IgIV, dexametasona, etopósido, PFC | Muerte a los 2 años de edad | |
| 2 a | F | 2 a | 2 a | Hemoglobina 6.5 g/L (11.5-14 g/dL), leucocitos 7140 cél/ μL (4000-10 000 cél/ μL), neutrófilos 6780 cél/μL (1500-6000 cél/μL), linfocitos 284 cél/μL (1500-6000 cél/μL), plaquetas 97 000 cél/μL (150- 450 000 cél/μL), fibrinógeno 120 mg/ dL (> 150 mg/dL), ferritina 1000 μg/L (< 300 μg/L), MO con hemofagocitosis, PCR complejo M. tuberculosis positiva 1,2,3DHR sin producción de radicales libres | Tuberculosis pulmonar. Oclusión intestinal con linfadenitis crónica granulomatosa |
Choque séptico. SDRA grave. Síndrome de activación macrofágica. Tuberculosis miliar |
Meropenem, vancomicina, fluconazol, IgIV, dexametasona | Muerte a los 2 años de edad | |
| Defectos en la inmunidad intrínseca e innata | 10 m | M | 4 m | 5 m | Leucocitos 32 800 cél/μL (4000- 10 000 cél/μL), Baja expresión de IL12RB1 | Absceso pulmonar lóbulo inferior izquierdo (salmonela grupo C | Secuestro pulmonar intralobar | Meropenem, vancomicina, ciprofloxacino, amikacina, IgIV | Vive |
| Desordenes autoinflamatorios | 4 a | M | 2 a | 3 a | PCR 7 a 24 mg/ dL (< 0.5 mg/dL), VSG 40 a 50 mm/h (< 20 mm/h), leucocitos 12 980-14 300 cél/μL (4000-10 000 cél/μL), plaquetas 478 a 523 000 cél/μL (150-450 000 cél/μL) | Fiebre periódica 1-2/mes de 1-3 días de duración, faringoamigdalitis recurrente exudativa, adenopatía cervical, aftas (2) | Sinusitis maxilar. Rinitis alérgica. IVU (E. coli) |
Múltiples antibióticos. Prednisolona en los ataques | Vive. Sin episodios actuales |
| 7 a | M | 3 a | 4 a | Leucocitos 13 a 19 980 cél/μL (4000-10 000 cél/ μL), PCR 3 a 10 mg/ dL (< 0.5 mg/dL), VSG 35 a 45 mm/h (< 20 mm/h) | Fiebre 2-3/ mes de 1-2 días de duración, faringoamigdalitis recurrente, adenopatía cervical bilateral | Rinitis alérgica | Múltiples antibióticos. Prednisolona en los ataques | Vive. Episodios esporádicos |
En la columna de infecciones y germen aislado, entre paréntesis se indica el número de episodios infecciosos cuando fue más de uno.
a = años; m = meses; d = días; IgIV = inmunoglobulina intravenosa; IVU = infección de vías urinarias; TCPH = trasplante de células progenitoras hematopoyéticas; MO = médula ósea; CsA = ciclosporina A; ITE = inmunoterapia alérgeno específica; IGSC = inmunoglobulina subcutánea; CsA = ciclosporina A; TMP-SMX = trimetoprima-sulfametoxazol; 1,2,3DHR = dihidrorodamina; CID = coagulación intravascular diseminada; PCV23 = vacuna polisacárido de neumococo de 23 serotipos; OMA = otitis media aguda; PCR = proteína C reactiva; VSG = velocidad de sedimentación globular; EIS = edad de inicio de síntomas; EAD = edad al diagnóstico; GEPI = gastroenteritis probablemente infecciosa.9
*Diagnóstico definitivo en los criterios bien establecidos para EII por la ESID (https://esid.org/Education/Diagnostic-Criteria-PID).9
Discusión
Se estima que el 70 % de los pacientes con EII no están siendo diagnosticados, lo que ha motivado la formación de equipos médicos y de diagnóstico especializados en EII (centros de diagnóstico para inmunodeficiencias Jeffrey Modell) y reuniones bianuales por comités de expertos de la IUIS para clasificar a los EII. Actualmente ya se ha encontrado más de 430 defectos monogénicos identificados. El registro nacional y regional de los EII es una herramienta útil para calcular su incidencia, prevalencia, distribución de frecuencia por regiones y países.12
Además de ESID y LASID, otros países se han agrupado pare realizar registros de los EII para conocer su epidemiología, características clínicas, tratamiento y herramientas para el diagnóstico. En el año 2016 países de Asia como Japón, Taiwán, Irán y Corea del Sur establecieron la Asia Pacific Society for Immunodificiencies, donde se destaca un incremento del registro y publicaciones de EII por:13
Mayor conocimiento de los EII por los médicos de primer contacto.
Mejores instalaciones para el diagnóstico y tratamiento como acceso a IGIV de reemplazo gratuita y TCPH.
Concientización de la población sobre EII y aporte de recursos óptimos a los centros de atención de EII.
Capacitación de pediatras en el área de EII.
La African Society for Immunodeficiencies fue creada en 2008 en Marruecos; con el tiempo ha incrementado el número de EII en su registro, dado que la mayoría de los países que la conforman tienen limitaciones para la realización de estudios de laboratorios básicos, así como las herramientas inmunológicas y genéticas especializadas, ellos han empleado alternativas para el diagnóstico definitivo en centros de investigación de otros países del primer mundo.14 Sugerimos que los diferentes médicos de países en vías en desarrollo como México establezcan redes de colaboración con laboratorios de investigación líderes en el estudio de los EII.
El INP es el centro nacional de referencia de EII del México. García et al. reportaron una descripción de un primer registro de 171 casos de EII en el INP durante el periodo comprendido entre 1970 y 2001.15
Desde que en 2009 se estableció el registro en línea en la plataforma de LASID y hasta julio del 2020, el INP es el centro mexicano de registro LASID con mayor número de casos de EII. con 497 (27.4 %) de un total de 1815 reportados en todo México. Los 1318 restantes están registrados en 45 centros hospitalarios de diferentes regiones de México. La plataforma de LASID en línea no permite el acceso a la información de otros centros (se requiere usuario y clave), por lo que desconocemos cuál es la distribución por edad, diagnóstico, edades y zona geográfica de los diferentes EII.
La comparación entre el registro realizado por García et al en 2001 y el registro en la plataforma en línea de LASID muestra el incremento en la detección de los EII en México, el cual se debe a la difusión de los EII a nivel nacional por diferentes mecanismos:
A médicos de diferentes entidades de la República Mexicana se les ha impartido un curso denominado “Curso-taller de infecciones recurrentes” en el que se exponen casos clínicos de pacientes con infecciones que son habituales en la práctica clínica, en los que se debe sospechar un EII.
Se imparte un congreso bienal de EII en México (MEXGID).
Se inició un curso de Alta Especialidad en Inmunodeficiencias avalado por la Universidad Nacional Autónoma de México en el INP.
El INP apoya con pruebas diagnósticas a casos sospechosos de EII procedentes de hospitales de segundo nivel.
Donación de reactivos e implementación de ensayo de DHR en diferentes hospitales públicos de México en donde no se cuenta con una prueba de tamiz para enfermedades genéticas congénitas.16
Publicaciones científicas de casos y series de casos de EII diagnosticados en México, en los que propone el uso de tamizaje neonatal para la IDCG, debido a su elevada mortalidad sin tratamiento, escalas para determinar el nivel de sospecha de EII, programas educativos para médicos de primer contacto con redes de referencia a centros especializados para diagnóstico genético.17-22
Colaboración científica en el campo de las EII con otros laboratorios de investigación internacionales.23,24,25
Conclusión
Presentamos un panorama del registro de los EII en el ámbito médico internacional. El general el conocimiento de las EII ha incrementado en el mundo y por tanto han surgido nuevas estrategias para registrar estos casos. En el HPS hubo un incrementó en el número de casos de EII diagnosticados y reportados, debido al trabajo en equipo del personal cada vez más capacitado. Presentamos el registro de EII en la plataforma de LASID de un centro pediátrico de México. Invitamos a que otros hospitales pediátricos sigan este modelo. En países donde existen limitaciones en los recursos y atención a la salud, es importante seguir registrando e incrementar el número de centros hospitalarios en LASID, lo cual permite conocer la prevalencia real de los EII y favorecer el desarrollo de políticas sanitarias para ofrecer las herramientas en el diagnóstico, gestión en la atención, tratamiento e investigación médica de los EII.

