











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Antecedentes
El síndrome linfoproliferativo autoinmune (ALP, autoimmune lymphoproliferative syndrome) o síndrome de Canale-Smith, es producido por un grupo heterogéneo de alteraciones en los genes que regulan el fenómeno de apoptosis; se caracteriza por la tríada clínica de enfermedad linfoproliferativa, citopenias autoinmunes y predisposición a malignidad.1 El ALPS es una inmunodeficiencia primaria clasificada en el grupo de trastornos genéticos de la regulación inmunológica.2
La descripción inicial de la enfermedad fue realizada en 1967 por Canale y Smith cuando reportaron cinco pacientes con linfadenopatía, esplenomegalia y citopenias autoinmunes, cuadro que simulaba linfoma maligno.3
Epidemiología
Se desconoce la incidencia y prevalencia del ALPS. Los casos estimados en todo el mundo superan varios cientos, pero el número no se ha definido en forma confiable. Probablemente numerosos casos de ALPS no se han diagnosticado debido a la expresión fenotípica variable y a la constelación de síntomas que se superponen con muchas otras afecciones, en particular el síndrome de Evans y otros trastornos linfoproliferativos.4,5
El ALPS es reportado en varios grupos raciales y étnicos. El predominio masculino fue sugerido previamente y se confirmó tanto en una cohorte francesa de ALPS como en una cohorte de ALPS de los Institutos Nacionales de la Salud (NIH, National Institutes of Health). En la cohorte francesa, en familias con mutaciones de FAS la probabilidad de que un hombre con la mutación de FAS tuviera ALPS sintomático fue de aproximadamente 75 % en comparación con 51 % de las mujeres. En forma similar, en la cohorte NIH, 69 % de los hombres comparado con 46 % de las mujeres con mutaciones de FAS desarrollaron características evidentes de ALPS. Además, la proporción de pacientes masculinos respecto a los femeninos con ALPS fue de 2.2 en la cohorte francesa y de 1.6 en la cohorte NIH.6,7,8,9,10
Patogénesis
El defecto de la apoptosis en los linfocitos humanos se identificó a partir de los trabajos del grupo de Fischer et al. (1995) y Drappa et al. (1996), donde se incluyen pacientes de la descripción original. El ALPS es la única enfermedad en humanos atribuida a un defecto primario en la muerte celular programada (apoptosis).8,9,11,12
La base molecular del ALPS consiste en la alteración de la apoptosis inducida por el receptor FAS, lo que provoca pérdida de la homeostasis linfocitaria. La mayoría de los pacientes tienen mutaciones heterocigotas en el gen FAS (TNFRSF6), pero también se han identificado mutaciones en el ligando de FAS y las caspasa (CASP) 10; al final, el defecto produce la acumulación crónica no maligna de células linfoides e incremento en la cuenta de linfocitos T CD3+ α/β+ CD4− CD8−, conocidos como células alfa/beta doble negativas (figura 1).1,7
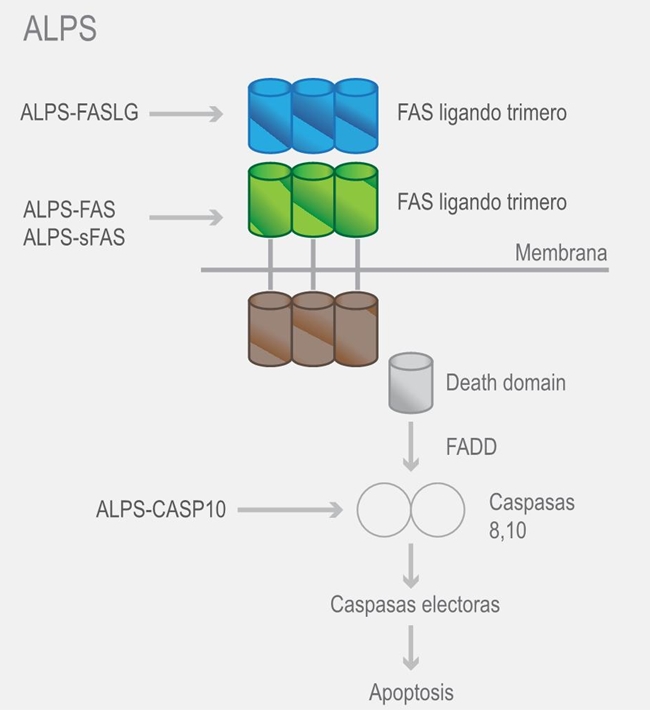
Figura 1 Vía de la apoptosis mediada por FAS y los sitios afectados en ALPS. ALPS-FASLG afectado en el trímero del ligando de FAS; ALPS-FAS y ALPS-sFAS en el trímero de FAS, ALPS-CASP10 en la caspasa 10. Death domain = dominio de muerte, FADD = FAS-associated death domain.
Cuando falla la vía extrínseca de la apoptosis mediada por la unión del ligando de FAS, se presenta el ALPS, que de acuerdo con la caracterización en el defecto molecular se ha clasificado como se muestra en el cuadro 1.13,14
Cuadro 1 Clasificación y frecuencia de las diferentes presentaciones de ALPS
|
Genotipo |
Fenotipo |
Defecto |
Frecuencia |
|
Mutaciones homocigotas del gen FAS también conocido como TNFRSF6 |
Prenatal, severo |
Ausencia total de FAS |
< 1 % |
|
Mutaciones heterocigotas del gen FAS también conocido como TNFRSF6 |
Infantes, moderado a severo con autoinmunidad |
Defecto funcional parcial de FAS |
79 % |
|
ALPS-sFAS |
Formas esporádicas |
Defecto funcional de FAS |
< 1 % |
|
ALPS-FASLG |
Mutaciones heterocigotas del gen de FASLG conocido como TNFSF6 |
Adultos, lupus eritematoso sistémico |
Ninguno |
< 1 % |
|
Mutaciones en |
Infantes, moderado con o sin autoinmunidad o inmunodeficiencia |
3 % |
||
ALPS-U |
Desconocido |
Infantes, moderado con autoinmunidad |
Desconocido |
17 % |
|
ALPS- |
Mutaciones en |
Infantes, moderado, autoinmunidad mínima con inmunodeficiencia, afectaciones cutáneas |
Defecto funcional de TNFRSF |
< 1 % |
ALPS PRCKδ |
Mutaciones en PRCKD |
Escolares, autoinmunidad, enfermedad cutánea lupus like, aumento IL-10 |
Defecto funcional de PKCD |
< 1 % |
|
(RALD) |
Infantes, autoinmunidad, ≥ 50 %, síndrome de Evans |
< 1 % |
ALPS = autoimmune lymphoproliferative syndrome, CEDS = ALPS-CASP8, FASLG = ligando de FAS, RALD = ALPS like-NRAS, IL = interleucina, TNFRSF = superfamilia de receptores de factor de necrosis tumoral, TNFSF6 = gen que codifica para FAS ligando, TNFRSF6 = gen que codifica para proteína FAS, CASP8 = gen que codifica para la caspasa 8, CASP10 = gen que codifica para la caspasa 10, NRAS = gen que codifica para proteína NRAS (GTPasa, oncogén, miembro de la familia de proteína RAS que ayuda a regular la división celular), KRAS = gen que codifica para proteína KRAS (GTPasa, oncogén, miembro de la familia de proteínas RAS que ayuda a la proliferación y maduración celular).
Se han sugerido más subclasificaciones, por ejemplo, para los defectos en la vía intrínseca de la apoptosis producidos por mutaciones en los genes NRAS y KRAS con cuadro clínico similar, sin embargo, actualmente se consideran como procesos asociados con ALPS.13,15
El gen FAS se localiza en el cromosoma 10q24.2 y consta de seis exones. Se han descrito más de 100 mutaciones heterocigotas dominantes de FAS (ALPS-FAS), dos terceras partes relacionadas con el dominio extracelular y el otro tercio, con el dominio intracelular; la mitad de las mutaciones resulta en modificación de la secuencia de aminoácidos y la otra mitad produce una proteína trunca. El patrón de herencia en la mayoría de los casos es autosómico dominante con penetrancia clínica incompleta, calculada en menos de 60 %.8,16
Las mutaciones heterocigotas en el gen FAS (ALPS-FAS) se han asociado con la expresión de moléculas mutantes de FAS, las cuales bloquean la señalización río abajo y no permiten la activación de las caspasas, es decir, tienen un efecto inhibitorio (dominancia negativa) que se traduce en deficiencia de apoptosis.10,17
Se describió la deficiencia del ligando de FAS (FASLG) en un adulto con cuadro clínico de lupus eritematoso sistémico con acentuada linfadenopatía sin esplenomegalia ni citopenias; el paciente resultó ser homocigoto para la mutación del gen, lo que sugiere un patrón dominante.18
La ausencia total de FAS (ALPS-FAS) se identificó inicialmente en un paciente de padres consanguíneos y posteriormente en dos individuos con linfadenopatía masiva desde el nacimiento, trombocitopenia autoinmune e infiltración pulmonar; este defecto es considerado causante de la forma más severa de ALPS.9,11,19,20,21
Se han descrito mutaciones en los genes que codifican para CASP10. La deficiencia de CASP8 se reportó en dos pacientes de una familia consanguínea con linfadenopatía masiva y con infecciones de repetición en tractos respiratorio superior e inferior, así como infecciones por herpes virus; el defecto en CASP8 se considera una entidad asociada con el ALPS o inmunodeficiencias con un fenotipo similar al de ALPS (ALPS-like).16,22,23
Manifestaciones clínicas
Aunque los fenotipos son variables, las manifestaciones clínicas por lo general se presentan en la infancia temprana, con media de edad de 24 meses y rango de 0 a 15 años, sin embargo, se han reportado pacientes con ALPS con inicio de manifestaciones clínicas más allá de los 17 años; incluso, se ha informado de manifestaciones desde la etapa en útero.24,25
Las principales manifestaciones clínicas son la tríada linfadenopatía, esplenomegalia y citopenias autoinmunes (cuadro 2). La linfadenopatía puede oscilar entre los límites normales para la edad hasta una linfadenopatía masiva, incluyendo masas torácicas y abdominales, situación que asociada a las citopenias autoinmunes provoca que estos pacientes sean referidos primariamente a los servicios de hematología y oncología.
Cuadro 2 Frecuencia de hallazgos clínicos en pacientes con ALPS
Hallazgo clínico |
% |
Acumulación linfoide |
100 |
Esplenomegalia |
90 |
Linfadenopatía |
87 |
Hepatomegalia |
45 |
Linfoma |
3 |
Anemia hemolítica autoinmune |
53 |
|
Púrpura trombocitopénica autoinmune |
44 |
Neutropenia autoinmune |
31 |
Urticaria |
13 |
Glomerulonefritis |
8 |
Hepatitis autoinmune |
6 |
Uveítis |
5 |
Todos los pacientes presentan esplenomegalia, linfadenopatía y la mayoría tiene manifestaciones de autoinmunidad, principalmente citopenias autoinmunes; otras manifestaciones son glomerulonefritis, hepatitis autoinmune, uveítis y vasculitis.26
Las citopenias autoinmunes generalmente son graves y difíciles de tratar y la lisis mediada por anticuerpos empeora por el hiperesplenismo con escasa respuesta al tratamiento habitual.27
Se ha observado que la linfoproliferación masiva tiende a mejorar con la edad, pero no así las manifestaciones de autoinmunidad en múltiples órganos, las cuales se incrementan durante el seguimiento a largo plazo.15
En lactantes, será importante distinguir las manifestaciones de ALPS de la infección por el virus de inmunodeficiencia humana. Cuando las citopenias autoinmunes afectan a más de un linaje celular, el ALPS se puede confundir con linfohistiocitosis hemofagocítica (linfadenopatía, pancitopenia, esplenomegalia e inflamación).28
Se ha demostrado que hasta 50 % de los pacientes con síndrome de Evans (caracterizado por citopenias autoinmunes) tiene hallazgos compatibles con ALPS.29,30
Los pacientes con ALPS tienen mayor susceptibilidad al desarrollo de malignidad (10 a 20 %), especialmente linfomas, lo mismo que los familiares portadores.26,31
Cuando además de linfadenopatía crónica no maligna hay infecciones severas recurrentes, generalmente por herpes virus o infecciones sinopulmonares, se debe pensar en deficiencia de CASP8.23
Aun cuando la elevación de linfocitos T dobles negativos (DNT) se considera un criterio requerido para el diagnóstico de ALPS, estos se han encontrado también elevados en pacientes con anemia aplásica, entre 4.3 y 9.1 % en sangre periférica. No está claro si la elevación de DNT participa en la fisiopatogenia del síndrome o si solo es un reflejo de la apoptosis deficiente durante el entrenamiento y selección en el timo, o de un descarrilamiento en la regulación inmunitaria que lleva a la autoinmunidad. Mientras que lo normal es encontrar menos de 1.5 a 2 % de DNT en sangre periférica, los pacientes con ALPS suelen tener 40 % o más. En todo caso, la elevación de DNT se comporta como un marcador altamente sensible pero inespecífico para ALPS.32
Los niveles elevados de vitamina B12 se correlacionan con niveles intracelulares de haptocorrina en los linfocitos de pacientes con ALPS. La cobalamina (vitamina B12) es esencial para la división celular; en el suero viaja acoplada a las proteínas transcobalamina y haptocorrina. Los linfocitos de pacientes con ALPS expresan aproximadamente 25 veces más haptocorrina que los sujetos sanos. Además, del ALPS se ha encontrado niveles séricos elevados de vitamina B12 en algunas neoplasias diseminadas, trastornos mieloproliferativos, síndrome hipereosinofílico, insuficiencia renal y hepatopatías.33
Las deficiencias de FADD (FAS-associated death domain, también conocido como MORT1) y CASP8 se superponen parcialmente con el ALPS-FAS (esplenomegalia, defecto de apoptosis inducido por FAS); se diferencian en presentar una deficiencia inmunológica en lugar de proliferación y autoinmunidad no controladas. Los pacientes que cumplen con los criterios de diagnóstico de ALPS con causa genética indeterminada se clasifican como ALPS-U (ALPS-unknown), lo que denota una categoría aún no definida. ALPS-U se presenta aproximadamente en 20 % de los pacientes con ALPS.34
Signos ausentes
En la historia familiar de los pacientes con ALPS no suele haber consanguinidad y en los antecedentes personales no hay dismorfias faciales, alteraciones óseas, malformaciones, retraso en el crecimiento o en el desarrollo, ni reacción adversa a la vacuna BCG; no suele haber infecciones recurrentes o graves, aunque sí infecciones asociadas con citopenias o tratamiento (esteroides, esplenectomía, rituximab, etcétera). En algunos pacientes se ha reportado susceptibilidad al virus de Epstein-Barr, que puede desencadenar linfomas. Los niveles séricos de inmunoglobulinas suelen ser normales o elevados. A pesar del defecto en la apoptosis, no se ha asociado ALPS con linfohistiocitosis hemofagocítica.
Diagnóstico (figura 2)

Figura 2 Algoritmo diagnóstico del síndrome linfoproliferativo autoinmune (ALPS, autoimmune lymphoproliferative syndrome). DNT = células T dobles negativas, IL = interleucina, FASL = ligando FAS, ALPS-FAS = ALPS debido a mutaciones de la línea germinal en el gen FAS, ALPS-sFAS = ALPS debido a mutaciones somáticas en el gen FAS, CASP10 = gen de la caspasa 10, ALPS-CASP10 = ALPS debido a mutaciones en CASP10, FASLG = gen para el ligando FAS, ALPS-FASLG = ALPS debido a mutaciones en FASLG, ALPS-U = ALPS debido a defecto genético desconocido. Adaptado de ©2019 UpToDate, Inc. Graphic 96876, versión 2.0.
De acuerdo con la clasificación del grupo de ALPS de los NIH inicialmente se propusieron los siguientes criterios diagnósticos obligatorios:
Linfoproliferación crónica no maligna.
DNT elevadas (en muestras de sangre periférica y tejido) (figura 3).
Evidencia in vitro de alteraciones de la apoptosis mediada por FAS.
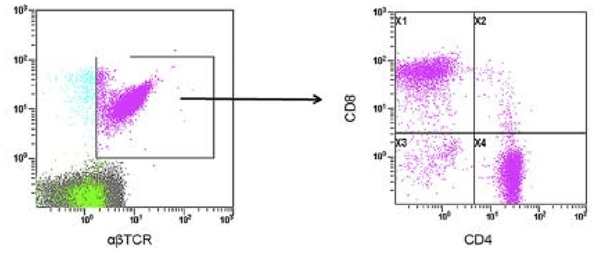
Figura 3 Células T dobles negativas (DNT) en células mononucleares de sangre periférica. La citometría de flujo de cuatro colores mostró mayor proporción (4.9 %) de células CD4CD8-DNT en la fracción αβTCR-CD3 (Gate X3). Tomado de Minemura et al.35
Otras anormalidades laboratoriales comúnmente encontradas son niveles elevados de IL-10 en sangre y aumento de los niveles de vitamina B12 en sangre periférica.6 Recientemente se ha demostrado que no todos los pacientes con ALPS cumplen con los criterios iniciales, por lo que se han desarrollado nuevos criterios diagnósticos (cuadro 3).13-14,30
Cuadro 3 Criterios diagnósticos para ALPS
Criterios requeridos |
1. Esplenomegalia y/o linfadenopatía crónica (> 6 meses) no malignas y no infecciosas |
2. Elevación marcada de DNT en sangre periférica (> 1.5 % del total de linfocitos o > 2.5 % del total de CD3) con base en cuentas normales o elevadas de linfocitos |
Criterios accesorios |
Primarios |
1. Alteración en la apoptosis de linfocitos in vitro (dos ensayos por separado) |
2. Mutaciones somáticas o de línea germinal en FAS, FASLG o CASP10 |
Secundarios |
1. Elevación de FASLG soluble en plasma o niveles elevados de IL-10 (> 200 pg/mL) o elevación de vitamina B12 plasmática (> 1500 ng/mL) o niveles elevados de IL-18 (> 500 pg/mL) |
2. Hallazgos inmunohistológicos característicos revisados por un patólogo experto |
3. Citopenias autoinmunes (trombocitopenia, neutropenia, anemia hemolítica, además de elevación de IgG sérica (policlonal) |
4. Historia familiar de linfoproliferación crónica no maligna, no infecciosa, con o sin autoinmunidad |
|
Diagnóstico definitivo: dos criterios requeridos + un criterio accesorio primario Diagnóstico probable: dos criterios requeridos + un criterio accesorio secundario |
ALPS = autoimmune lymphoproliferative syndrome, CASP = caspasa, FASLG = ligando de FAS, DNT = células T dobles negativas, IgG = inmunoglobulina G.
Varias observaciones sugieren que eventos genéticos adicionales llevan a enfermedad proliferativa indolente, principalmente del compartimiento de linfocitos, o a leucemia agresiva. La identificación de estos factores sería de gran interés para el tratamiento terapéutico de los pacientes con mutaciones somáticas de FAS.32,33
Diagnóstico diferencial
Otras patologías que pueden tener algunas manifestaciones clínicas similares al ALPS son enfermedad de Castleman, enfermedad de Rosai-Dorfman, síndrome linfoproliferativo ligado al cromosoma X, enfermedad linfoproliferativa y autoinmune de Dianzani y enfermedad de Kikuchi-Fujimoto; debido a lo anterior, se deben incluir dentro del diagnóstico diferencial.28,29,30
Las inmunodeficiencias primarias por ganancia de función (GOF) de STAT1 y STAT3, y la deficiencia de STAT5B cursan también con linfoproliferación, sin embargo, en STAT5B y STAT3-GOF hay retraso considerable en el crecimiento; en STAT1-GOF suele haber infecciones por virus, hongos y micobacterias. Asimismo, se debe considerar cualquier otra inmunodeficiencia por desregulación inmunitaria que curse con linfoproliferación o autoinmunidad, incluyendo APDS-PASLI.
Se han descrito varias inmunodeficiencias ALPS-like incluyendo deficiencia de ADA2, deficiencia de LRBA, deficiencia de MST1 por variantes patogénicas en STK4 y haploinsuficiencia de TNFAIP3.35,36,37,38,39,40
Tratamiento
La determinación de la etiología genética es de particular importancia para el tratamiento de los pacientes con ALPS. Los fármacos citotóxicos o citostáticos, incluidos el metotrexato, la mercaptopurina y sus derivaciones o el micofenolato de mofetilo, han demostrado gran eficacia en pacientes con ALPS-FAS, sin embargo, pueden tener una toxicidad importante a largo plazo. Con base en experimentos en ratones y la demostración de la hiperactivación de la vía mTOR (mechanistic target of rapamycin) en las células T ALPS-FAS, se demostró que los inhibidores de mTOR tienen gran eficacia con efectos secundarios mínimos en cohortes de pacientes con ALPS-FAS.33,34,35,41
Metas del tratamiento
El tratamiento de ALPS se centra en tres aspectos: tratamiento de las manifestaciones de la enfermedad, tratamiento y prevención de las complicaciones derivadas de la enfermedad y tratamiento curativo. La mayoría de la experiencia se limita a los pacientes con ALPS-FAS, en quienes la enfermedad linfoproliferativa comienza a temprana edad y tiende a hacerse menos frecuente en la adolescencia y en la edad adulta. El desarrollo de enfermedad autoinmune cambia el tratamiento y el pronóstico.42,43
Evaluación y seguimiento del diagnóstico inicial
La presencia y el alcance de la linfoproliferación o la autoinmunidad deben determinarse antes de iniciar el tratamiento en una persona recién diagnosticada con ALPS o con sospecha de este síndrome, lo cual se puede lograr con una combinación de examen físico, estudios de imágenes (por ejemplo, tomografía computarizada o tomografía por emisión de positrones) de áreas del cuerpo que se cree están involucradas (típicamente cuello, tórax, abdomen y pelvis) y una evaluación de laboratorio que incluya la cuantificación de DNT circulantes, niveles de biomarcadores séricos (por ejemplo, vitamina B12, IL-10, FASLG), hemograma y naturaleza de los autoanticuerpos (por ejemplo, dirigidos contra las células sanguíneas).
En los pacientes que se considere necesario el tratamiento inmunosupresor, también se debe iniciar una investigación básica de la integridad del sistema inmunológico, particularmente en pacientes jóvenes. Estos estudios incluyen hemograma completo con niveles de inmunoglobulina, títulos de anticuerpos a proteínas (y, si tienen menos de dos años, polisacáridos), vacunas y CH50, así como subpoblaciones de linfocitos y estudios funcionales de las células T si se considera el tratamiento a largo plazo (por ejemplo con sirolimus).
Debe incluirse evaluación para linfoma en pacientes con linfadenopatía extensa, especialmente en quienes tienen síntomas inespecíficos (por ejemplo, fiebre, pérdida de peso, sudores nocturnos). Las imágenes de la tomografía por emisión de positrones pueden ser útiles en este sentido, ya que a menudo es difícil distinguir entre linfadenopatía benigna (ALPS específica) y linfoma maligno, que puede presentarse concomitantemente. La tomografía por emisión de positrones puede dirigir al cirujano hacia los ganglios linfáticos con alta actividad metabólica, que tienen mayor probabilidad de utilidad en el diagnóstico. En la investigación del linfoma es particularmente importante considerar los agentes inmunosupresores o inmunomoduladores, cuyo uso puede alterar o enmascarar el desarrollo del linfoma.
Existe evidencia de que la vigilancia periódica mediante imágenes anatómicas combinadas, es decir, tomografía computarizada o por emisión de positrones, puede ser útil para monitorear la eficacia y el tiempo necesario de administración del sirolimus, aunque es indispensable la recopilación de datos a largo plazo para determinar el costo-beneficio de estos estudios de imagen antes de que se usen de forma rutinaria.15,44,45,46,47,48
Diversos hallazgos de laboratorio, incluido el número de DNT y los niveles de IL-10, FASLG y otros biomarcadores, no solo tienen un propósito diagnóstico, sino que también se pueden usar para monitorear la actividad de la enfermedad y la respuesta al tratamiento.
Esta evaluación ayuda a definir con qué agente o agentes se tratará al paciente. Si se toma la decisión de no iniciar el tratamiento, se recomienda un seguimiento periódico con los recursos descritos.
Tratamiento de las manifestaciones clínicas de la enfermedad
El tratamiento de las manifestaciones de la enfermedad se centra en el control o tratamiento de la linfoproliferación, autoinmunidad y del linfoma. El tratamiento de la linfoproliferación y la enfermedad autoinmune en el ALPS se basa en los datos de observación y la experiencia clínica, ya que no hay ensayos aleatorios. El tratamiento es individualizado y la elección del agente depende de diversos factores específicos del paciente y de la preferencia y experiencia del médico tratante.15,42,45,46,47,48,49
Una vez iniciado, el tratamiento inmunosupresor o inmunomodulador puede ser difícil de suspender, lo que se debe tener en cuenta debido a los efectos secundarios, los riesgos y el costo a largo plazo. El desarrollo de citopenias autoinmunes sintomáticas a menudo indica una nueva fase en el curso del ALPS. Los tratamientos que afectan el sistema inmunológico no se pueden usar durante largos periodos (años), especialmente en niños, sin complicaciones. Por ejemplo, el uso a largo plazo de glucocorticoides se asocia con obesidad, retraso del crecimiento, hipertensión, cataratas, osteopenia-osteoporosis que conduce a fracturas óseas patológicas y diabetes, entre otros. El uso a largo plazo de agentes antiproliferativos de células T (por ejemplo, ciclosporina o tacrolimus) puede causar infecciones significativas e insuficiencia renal. Además, el sistema inmunológico puede no tolerar la supresión durante periodos prolongados. La resolución de las manifestaciones de ALPS después de años de tratamiento puede deberse a la inmunosupresión crónica en lugar de la remisión permanente de ALPS, lo que hace necesaria una evaluación periódica de la competencia inmunológica general. Lo anterior es particularmente importante en pacientes jóvenes, ya que experimentan y adquieren infecciones virales comunes, como las causadas por el citomegalovirus y el virus de Epstein-Barr.
Linfoproliferación
Se sugiere el uso de tratamiento inmunosupresor solo para complicaciones graves de la linfoproliferación (por ejemplo, esplenomegalia masiva u obstrucción de las vías respiratorias por agrandamiento del tejido amigdalino o adenopatía cervical) o en presencia de manifestaciones autoinmunes concomitantes que requieran tratamiento. No se recomienda la amigdalectomía para el tejido amigdalino agrandado que obstruye la vía aérea porque puede reaparecer.
Las manifestaciones de la linfoproliferación se pueden suprimir mediante agentes inmunosupresores como glucocorticoides, ciclosporina, sirolimus, tacrolimus o micofenolato de mofetilo. Generalmente no es necesaria la inmunosupresión para la linfoproliferación en ausencia de autoinmunidad, ya que sus efectos secundarios contrapesan sus beneficios. Además, la linfadenopatía, la esplenomegalia y la hepatomegalia se manifiestan de nuevo una vez que se suspende la inmunosupresión.8,50,51,52,53
El micofenolato de mofetilo es generalmente bien tolerado y requiere poco monitoreo, por lo que suele ser el agente de elección con el cual comenzar el tratamiento. Parece inducir una modesta mejoría en la linfadenopatía y cierta mejoría en la esplenomegalia (en algunos pacientes) y marcadores de laboratorio, como los recuentos de linfocitos y los niveles de IL-10 y vitamina B12. La dosis y duración del tratamiento deben ser individualizadas para cada paciente. Se debe determinar una dosis mínima que cumpla con los objetivos terapéuticos.15,53,54,55
La experiencia con sirolimus sugiere que puede afectar la linfoproliferación más sostenidamente que otros agentes inmunosupresores, como lo demuestra una reducción más significativa en la adenopatía, la esplenomegalia y los biomarcadores (por ejemplo, el receptor de IL-2 soluble alfa, FAS ligando soluble, IL-10, IL-18 y vitamina B12). El aumento en la activación de la vía de la rapamicina (mTOR) en ALPS puede proporcionar una explicación para la acción favorable de sirolimus. Sin embargo, esto se ve compensado por el hecho de que el sirolimus requiere monitoreo de niveles, efectos secundarios y complicaciones, incluidas infecciones. Por otro lado, es menos mutagénico que el micofenolato de mofetilo, tacrolimus y ciclosporina. Además, el uso prolongado de los inhibidores de la calcineurina (por ejemplo, tacrolimus y ciclosporina) se asocia con toxicidad renal.53,54,55,56
Primero se debe determinar si el tratamiento es necesario, con base en las consideraciones expuestas. De ser así, se debe comenzar con el agente menos inmunosupresor para lograr los objetivos terapéuticos, especialmente si se necesita una inmunosupresión más allá de algunas semanas. En la mayoría de los pacientes, esto se puede lograr con 300 a 600 mg/m2/día de micofenolato de mofetilo, con o sin un ciclo corto (aproximadamente de dos a tres semanas) de prednisona. Generalmente se utiliza prednisona de dos a tres semanas, dosificada entre 1 y 2 mg/kg/día, con disminución progresiva según las indicaciones de la respuesta clínica o los efectos secundarios, incluida la tolerabilidad del agente, el examen físico (por ejemplo, grado de la esplenomegalia) y biomarcadores para determinar la respuesta y guiar los cambios en la dosis del fármaco. Puede ser difícil distinguir entre la mejoría espontánea de la linfoproliferación y el efecto de los fármacos, particularmente en pacientes < 10 años. Por lo tanto, se sugiere una disminución progresiva de los medicamentos. En pacientes con respuesta insuficiente al micofenolato, autoinmunidad concomitante (o nueva) o intolerancia al micofenolato, se inicia el sirolimus con una dosis que mantenga los niveles más bajos del fármaco dentro del rango terapéutico (generalmente 2 a 3 mg/m2/día).57,58
La esplenectomía no se recomienda en pacientes con ALPS, debido a la falta información a largo plazo de beneficio terapéutico sostenido y al aumento de la incidencia de sepsis posesplenectomía, tanto en niños como en adultos. En la cohorte francesa de ALPS, 33 % de 90 pacientes fue sometido a esplenectomía (40 % por esplenomegalia masiva y 60 % por citopenias autoinmunes resistentes). En la cohorte NIH, 44 % de 150 pacientes necesitaron esplenectomía, tres cuartas partes por citopenias resistentes que típicamente involucraban más de un linaje. De los pacientes que tuvieron esplenectomía por citopenias severas, 50 % en la cohorte francesa recidivó y en la cohorte NIH más de 70 % recayó a los cuatro y 20 años de la esplenectomía. La trombocitopenia fue la citopenia más frecuente en los pacientes que recidivaron.58,59
También se observó aumento de la tasa de sepsis en los pacientes sometidos a esplenectomía en ambas cohortes. En la francesa, nueve de 30 pacientes sufrieron 17 episodios de infecciones bacterianas invasivas graves, con cuatro muertes; 27 de 66 en la cohorte del NIH sufrieron uno o más episodios de sepsis, con siete muertes. Se descubrió que la edad temprana es un factor de riesgo para la sepsis posterior a la esplenectomía, sin que la profilaxis antimicrobiana y las vacunaciones adecuadas la impidan.60,61
Manifestaciones autoinmunes
Las manifestaciones autoinmunes suelen responder a los agentes inmunosupresores. El desarrollo de manifestaciones autoinmunes, que invariablemente involucran combinaciones de las citopenias autoinmunes (anemia hemolítica autoinmune y trombocitopenia con o sin neutropenia), señala una transición en la historia natural del ALPS, que a menudo se caracteriza por la necesidad de iniciar o escalar el tratamiento. Los pacientes con manifestaciones autoinmunes suelen tener más dificultades para abandonar la tratamiento inmunosupresor que los pacientes solo con linfoproliferación.34,49,60,61,62,63,64
Los glucocorticoides administrados por vía oral o en bolos intravenosos pueden ser efectivos en episodios agudos y graves de anemia o trombocitopenia. La transición a los agentes ahorradores de esteroides se realiza a medida que las dosis de glucocorticoides se reducen. Aproximadamente entre 50 y 60 % de los pacientes requieren fármacos inmunosupresores adicionales para controlar la autoinmunidad debido a respuestas incompletas a los glucocorticoides solos.
El micofenolato de mofetilo, la ciclosporina, el tacrolimus y particularmente el sirolimus son efectivos en las citopenias autoinmunes crónicas resistentes y pueden permitir la disminución progresiva de los glucocorticoides o evitarlos. Un estudio prospectivo multiinstitucional demostró la eficacia de la monoterapia con sirolimus en ALPS; los pacientes lograron una respuesta completa y duradera, que incluyó mejoría en la enfermedad autoinmune, linfadenopatía y esplenomegalia en uno a tres meses desde el inicio del sirolimus.34,43,62,63,64,65,66,67,68
Si bien el rituximab no es la primera opción, se ha utilizado con éxito en el tratamiento de las citopenias resistentes a ALPS, aunque todavía no se sabe cuánto tiempo los individuos afectados permanecerán en remisión clínica. La experiencia sugiere que el monitoreo de las subpoblaciones de células B puede ayudar a estimar la duración de la remisión y también a determinar la posible hipogammaglobulinemia posterior a la administración de rituximab. En pacientes asplénicos con ALPS, los efectos adversos relacionados con el fármaco, incluida hipogammaglobulinemia y neutropenia inducida, pueden aumentar el riesgo de infección.15,63,68,69,70,71
La inmunoglobulina intravenosa (IVIG) en dosis altas es suficiente para controlar las citopenias en algunos pacientes. Las citopenias graves (resistentes) u otras manifestaciones autoinmunes potencialmente mortales pueden requerir el uso de plasmaféresis en combinación con otras modalidades, incluido el bortezomib.49,72 Por lo general se emplea una combinación de agentes inmunosupresores e inmunomoduladores. Dependiendo de la gravedad, se puede utilizar una dosis alta de IVIG (para inmunomodulación a razón de 1 a 2 g/kg), un pulso de prednisona y el inicio de micofenolato; si es necesario, se debe cambiar a un agente más potente (por ejemplo, de micofenolato a sirolimus o tacrolimus). Si la IVIG en dosis altas no logra la remisión o no la mantiene (lo que comúnmente sucede en ALPS), se suele utilizar rituximab como agente inmunomodulador, teniendo en cuenta que el paciente puede necesitar un reemplazo de inmunoglobulina secundario al desarrollo de hipogammaglobulinemia.
El linfoma se trata de acuerdo con los protocolos convencionales. La apoptosis mediada por FAS defectuosa no impide la respuesta a los agentes quimioterapéuticos o la radiación.
El rituximab y los inhibidores de la rapamacina, como el sirolimus y la pentostatina, se han aplicado en pacientes con enfermedad resistente o considerable linfoproliferación; han sido efectivos en la remisión de citopenias y linfadenopatías.15,43,44,45
La esplenectomía está indicada en citopenias resistentes, especialmente en anemia hemolítica y trombocitopenia autoinmune, sin embargo, se sugiere como última alternativa, principalmente por el riesgo posterior de infecciones graves y sepsis.32,42
El trasplante alogénico de células progenitoras hematopoyéticas es el tratamiento curativo y se ha indicado en pacientes con enfermedad grave o resistente.55,56
Prevención de complicaciones
La mayoría de las complicaciones se deben a los agentes inmunosupresores e inmunomoduladores y se relacionan con riesgo de infecciones o efectos secundarios sistémicos de los glucocorticoides. El uso de inmunoglobulina intravenosa o subcutánea está justificado en algunos casos, así como de antimicrobianos para prevenir infecciones oportunistas, fúngicas y virales. Las infecciones causadas por agentes inmunosupresores e inmunomoduladores deben investigarse exhaustivamente con cultivos y tratarse agresivamente.
En los pacientes que han tenido esplenectomía, se recomienda la profilaxis con penicilina de por vida y la posterior vacunación de refuerzo.
Tratamiento curativo
El trasplante de células progenitoras hematopoyéticas (TCPH) es el único tratamiento curativo para el ALPS. Los regímenes de preparación de intensidad reducida ocasionan menor morbilidad y mortalidad que los regímenes de preparación mieloablativa; se han utilizado con éxito en las inmunodeficiencias primarias. El protocolo de acondicionamiento propuesto por Bleesing et al. incluye alemtuzumab, fludarabina y melfalán como esquema bien tolerado de intensidad reducida. El estado de la actividad del ALPS debe evaluarse cuidadosamente y las posibles consecuencias de los tratamientos a largo plazo, incluida la función del órgano, que deben compararse con los riesgos del trasplante.56,73,74
Indicaciones del TCPH
Desarrollo de linfoma debido a recaída o segundo linfoma primario después del tratamiento.
Citopenias autoinmunes severas y resistentes, con manifestaciones de la enfermedad que solo pueden controlarse mediante una inmunosupresión agresiva y sostenida.
Desarrollo de un trastorno de inmunodeficiencia significativo durante o después de inmunosupresión prolongada.
Fenotipo de enfermedad grave en el momento del diagnóstico debido a mutación homocigota y heterocigota compuestas de FAS.
Defecto genético adicional que complique el manejo.
Infección bacteriana invasiva recurrente grave o sepsis después de una esplenectomía, a pesar de una adecuada profilaxis antimicrobiana y vacunas adecuadas.56,74
Consejo genético
Se recomienda la asesoría genética para proporcionar a los pacientes y las familias información sobre la naturaleza de la enfermedad, su herencia y las implicaciones para los familiares y portadores afectados. Los pacientes con ALPS debido a mutaciones somáticas de FAS (ALPS-sFAS) no heredaron el defecto genético de ninguno de los padres y no parecen tener la mutación en el ADN de línea germinal; en consecuencia, su descendencia no heredará la mutación somática. En contraste, quienes tienen una mutación constitucional tienen un riesgo de 50 % de transmitir la mutación a cada uno de sus descendientes, quienes pueden verse afectados en mayor o menor medida debido a la penetrancia variable y expresividad de las manifestaciones de ALPS.
Pronóstico
Muchos pacientes con ALPS tienen un pronóstico favorable, la linfadenopatía disminuye con el tiempo y las características autoinmunes permanecen ausentes o manejables con necesidad limitada de tratamiento inmunosupresor. Algunos pacientes con mutaciones particulares que afectan el dominio de muerte intracelular de la proteína de FAS pueden tener un peor pronóstico, mientras que aquellos con defectos en el dominio extracelular pueden tener una enfermedad más leve. Sin embargo, es imposible predecir qué camino seguirá cada paciente, independientemente del genotipo. Por lo tanto, el seguimiento a largo plazo está indicado; cada paciente debe ser monitoreado cuidadosamente durante el curso de la enfermedad.
Componentes importantes del seguimiento son la educación y el empoderamiento de los pacientes y sus familias. Además, es posible que sea necesario el monitoreo para detectar eventos patógenos posteriores que afecten la apoptosis mediada por FAS, la homeostasis de los linfocitos en general u otros eventos (aún desconocidos). La tasa de supervivencia a 50 años se ha estimado en 85 % para los pacientes con deficiencia de FAS. Las principales causas de muerte en ALPS son sepsis posterior a esplenectomía y el desarrollo de linfoma. La vigilancia para detección de linfoma debería incluir monitoreo periódico del virus de Epstein-Barr en suero, ya que se ha reportado viremia progresiva como predictor.53,75,76,77
Mensajes clave
El ALPS es un trastorno de la regulación inmunológica provocado por alteraciones genéticas que afectan el fenómeno de la apoptosis de los linfocitos y se caracteriza por la desregulación del sistema inmunitario.
Los genes afectados en el ALPS son FAS o TNFRSF6, FASLG o TNFSF6, CASP10, PRCKD.
Existen padecimientos con manifestaciones similares con mutaciones en los genes CASP8, KRAS y NRAS.
El ALPS es una enfermedad rara, subdiagnosticada y de la cual se desconoce su incidencia, pero que por sus manifestaciones clínicas puede confundirse con otras patologías, especialmente hematooncológicas.
ALPS-sFAS y ALPS-FAS son los tipos más comunes de ALPS y son causados por mutaciones somáticas y de línea germinal, respectivamente, en el gen FAS. En casos raros, el ALPS está causado por mutaciones en los genes que codifican el ligando FAS (ALPS-FASLG) o CASP10 (ALPS-CASP10).
El fenotipo de ALPS resulta de la incapacidad para regular la homeostasis de los linfocitos debido a apoptosis defectuosa mediada por la vía del ligando FAS (FASLG). Los defectos en esta vía conducen a la expansión de poblaciones de linfocitos específicos de antígeno. FAS también parece desempeñar un papel en la supresión de la transformación maligna de los linfocitos.
Las características clínicas del ALPS son enfermedad linfoproliferativa crónica no maligna, citopenias autoinmunes y predisposición a malignidad.
El tratamiento del ALPS está encaminado a inducir la remisión del proceso autoinmune y la linfadenopatía mediante el uso de esteroides e inmunosupresores.
El tratamiento curativo es el trasplante de células progenitoras hematopoyéticas, especialmente en los casos graves. El pronóstico a largo plazo es bueno en general, con el riesgo latente del desarrollo de linfomas, sin embargo, empobrece cuando se elimina el bazo.
Algunas preguntas por responder en la próxima década son la utilidad diagnóstica de los niveles de vitamina B12 y DNT, así como el significado de la elevación de estos últimos.
Actualmente en el Instituto Nacional de Pediatría, en el Servicio de Inmunología y en la Unidad de Investigación de Inmunodeficiencias (Centro de Diagnóstico Jeffrey Modell), se realiza el diagnóstico y tratamiento de pacientes con sospecha de ALPS.

