











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Abreviaturas y siglas
BAFFR, B cell-activating factor receptor
ICOS, inductor de coestimulación
IDCV, inmunodeficiencia común variable
Ig, inmunoglobulina
TLR, receptores tipo Toll
Antecedentes
La inmunodeficiencia común variable (IDCV) es la inmunodeficiencia sintomática más común en la edad adulta. Tiene una prevalencia de 1 en 25 000-50 000 en la población general y una presentación bimodal, el primer pico entre los seis y 10 años y el segundo entre los 18 y 25 años de edad; con frecuencia, los pacientes presentan hasta siete años de retraso en el diagnóstico a partir del inicio de los síntomas.1,2,3
El diagnóstico de IDCV es de exclusión y debe considerarse en pacientes de cualquier edad que presenten hipogammaglobulinemia sin causa conocida.
Los criterios diagnósticos actuales de IDCV fueron plasmados en un Consenso Internacional realizado en 2016, con el fin de uniformar criterios, debido a la variedad de manifestaciones clínicas y anormalidades de laboratorio en estos pacientes.3,4,5
Para establecer el diagnóstico de IDCV, los pacientes deben presentar al menos infección, autoinmunidad o linfoproliferación, además de cumplir con los siguientes criterios:
Hipogammaglobulinemia, de acuerdo con el rango de referencia para la edad y el laboratorio donde se procesó la muestra, en al menos dos determinaciones, con un promedio de tres semanas de diferencia entre cada una. Las concentraciones de IgA o IgM deben estar al menos una desviación estándar por debajo del valor considerado de referencia para la edad.
En pacientes con concentraciones séricas de IgG > 100 mg/dL se recomienda evaluar las respuestas a antígenos dependientes e independientes de linfocitos T, en busca de alteración en al menos un tipo de antígeno.
Exclusión de causas secundarias de hipogammaglobulinemia.
Los estudios genéticos se requieren solo en los pacientes que presentan complicaciones, ya que la presencia de defectos genéticos únicos puede convertirlos en candidatos a terapias específicas.6
Células B de memoria
Posterior al desarrollo, independientemente de antígeno en la médula ósea, las células B inmaduras abandonan esta y se reúnen en el pool de células B maduras de larga vida, las células naive CD27−IgD+IgM+.
Cuando estas células son estimuladas por un antígeno, en presencia de una coestimulación adecuada, participan en una reacción en el centro germinal y posteriormente se transforman en células plasmáticas o células B de memoria (50 %).
Un importante número de pacientes con IDCV muestran alteraciones en el desarrollo de los linfocitos B, tanto células plasmáticas como de memoria, mientras que las células B maduras están presentes en número normal, lo que sugiere defectos en la diferenciación tardía de células B.7 Recordemos que la maduración de las células B es un proceso que inicia en la médula ósea y continúa en los órganos linfoides periféricos.
Las células pro-B derivadas de la médula ósea (CD19−CD10+/−CD20−CD22+CD24− vpreB-Igα+/−) se diferencian a células pre-B (CD19+CD10+CD20-CD24+verb+Igα+ intracelularμ+) y posteriormente a células B inmaduras/transicionales (CD19+CD10+ D20 CD24++IgM+).
Solo 10 a 20 % de estas células B transicionales abandonan la médula ósea, convirtiéndose en células B de transición tipo 1, T1 (IgMhi IgD−CD21−CD23+) y células B de transición tipo 2 (T2) (IgMhi IgD+CD21int CD23+). Estas células B transicionales, pueden convertirse en:
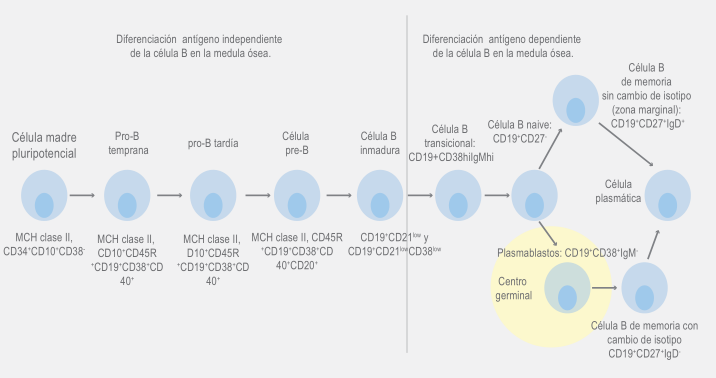
Aproximadamente 15 a 55 % de las células B circulantes son CD27+, de las cuales 50 % expresa IgM e IgD, denominadas células B de memoria sin cambio de isotipo y el resto con IgM−IgD− se denomina células B de memoria con cambio de isotipo.
Alrededor de 90 % de los pacientes con IDCV muestra un número células B normales, 5 a 10 % muestra reducción y solo 1 %, ausencia de estas.9
La ausencia de células B de memoria sugiere una reacción germinal insuficiente que puede asociarse con bloqueo de la transición de células T1 a T2 en pacientes con IDCV, debido a deficiencia del receptor BAFF (factor activador de linfocitos B).
Determinar la presencia y tipo de las células B transicionales en los pacientes con IDCV constituye un factor pronóstico de la enfermedad. El incremento de las células B transicionales se asocia con linfadenopatía; en contraparte, el aumento de las células B CD21low se relaciona con esplenomegalia, enfermedad granulomatosa y pronóstico pobre; estos datos muestran la importancia de determinar su presencia y tipo en los pacientes.
Otras alteraciones de los anticuerpos generados por las células B en los pacientes con IDCV son reordenamientos anormales en VDJ y en la región determinante 3 (CDR3), generando mayor diversidad de la célula B y disminución de la hipermutación somática en los repertorios de memoria. Estos cambios ocurren en etapas tempranas de la maduración (células pro-B) y podrían explicar el incremento de la autorreactividad, inmunodeficiencia y linfomas que presentan estos pacientes.10,11
Por otra parte, se ha estudiado la estimulación de las células B mediante la respuesta independiente del antígeno T. Las células dendríticas plasmacitoides pueden inducir diferenciación de las células B mediante los TLR (receptores tipo Toll) y citocinas como BAFF, APRIL (ligando inductor de proliferación), los ligandos de proteínas TACI (activador transmembrana y modulador de ligando de calcio, ciclofilina) y B cell-activating factor receptor (BAFFR).
La implicación de las vías de los TLR tiene sustento, ya que los defectos genéticos en su señalización conducen a mayor susceptibilidad de infecciones bacterianas y mala respuesta de anticuerpos secundaria a la pobre diferenciación de las células B.12,13
Los pacientes con IDCV presentan, además, defectos en las vías de señalización por alteración de las citocinas coestimuladores como BAFF, APRIL, los ligandos de proteínas TACI y BAFFR, alteraciones que se asocian con mayor susceptibilidad de infecciones bacterianas y mala respuesta de anticuerpos secundarios:
La presencia de mutaciones en ICOS (inductor de coestimulación).
Otro mecanismo que explica las alteraciones en la diferenciación de la célula B ocurre a través de las células T activadas, cuya interacción con el ligando ICOSL (expresado en la célula B) es esencial para formación del centro germinal y maduración de la célula B.
En consecuencia, los pacientes con IDCV presentan múltiples mecanismos que llevan a defectos en las células B, lo que se traduce en predisposión a autoinmunidad, linfoproliferación y formación de granulomas.14,15
Clasificación IDCV
Inicialmente la clasificación de acuerdo con el fenotipo de las células B establecía tres categorías de IDCV de acuerdo con la producción de anticuerpos in vitro:16
En 2002 se realizó la clasificación de Warnatz, que utiliza marcadores de memoria presentes en células B como CD27, CD21 y CD19:
Grupo 1a: Presenta porcentajes muy bajos de células B de memoria con cambio de isotipo; 100 % de los pacientes presenta esplenomegalia y 60 % manifiesta citopenia autoinmune, con alteraciones en el centro germinal.
Grupo 1b: Solo 7.7 % muestra asociación con vitiligo y anemia perniciosa.
Grupo II: Los pacientes pueden cursar con incremento en la proliferación o decremento en la apoptosis. Presentan alteración en la producción de anticuerpos in vivo y la hipogammaglobulinemia puede ser secundaria a fallas en la diferenciación hacia células plasmáticas.7,11,17
Posteriormente, un grupo alemán estableció la clasificación Freiburg para pacientes con IDCV, que permite distinguir pacientes con diferenciación alterada de las células B de memoria dependiente de alteraciones en el centro germinal con defectos en la diferenciación temprana de células B preváricas. Agrupa a los pacientes en tres grupos:
-
Tipo I < 0.4 % de linfocitos B CD27+IgM−IgD−.
Tipo II > 0.4 % de células B de memoria con cambio de isotipo.
Los pacientes portadores de IDCV ubicados en el grupo Ia muestran mayor prevalencia de cipolinas autoinmunes y esplenomegalia.11,18,19
Piqueras et al. propusieron la clasificación de París, acorde con la presencia de células B de memoria, formando tres grupos:
Grupo MB0, con prácticamente ausencia de células B de memoria.
Grupo MB1, con defecto en células B de memoria con cambio de isotipo, pero valores normales de células B de memoria sin cambio de isotipo.
Grupo MB2, con células B de memoria normales,
La última clasificación tuvo menos impacto como factor predictivo de complicaciones clínicas en pacientes con IDCV, por lo que no se utilizó de forma global.20
Las clasificaciones anteriores fueron empleadas para elaborar EUROclass, en un intento por unificar criterios en los pacientes con IDCV (Figura 2).

La clasificación EUROclass requiere la medición de los siguientes subtipos: células B totales, células B de memoria IgD−IgM−, células B transicionales y células B CD21low. Establece dos grupos principales:
-
I . El grupo B+ con más de 1 % de células B.
smB+ con más de 2 % de células B de memoria con cambio de isotipo:
smB+ 21low: células CD21 > 10 %
smB+ 21norm: células CD21low < 10 %
smB− con menos de 2 % de células B de memoria con cambio de isotipo.
smB-Trhi con más de 9 % de células B transicionales.
smB-Trnorm, que presentan menos del 9 % de células B transicionales
II. El grupo B−, con menos de 1 % de células B.
El grupo smB− Trhi fue el más asociado con linfadenopatías, mientras que el grupo smB+ 21low se asoció con mayor predisposición a esplenomegalia y linfoproliferación. Esta clasificación mostró mejor capacidad de pronosticar la presencia de linfadenopatías, enfermedad granulomatosa y esplenomegalia en comparación con las clasificaciones previas. Sin embargo, como el resto, no es del todo fiable para predecir autoinmunidad en pacientes con IDCV21,22,23 (Cuadro 1).
Cuadro 1 Comparación de diferentes clasificaciones de IDCV
| Clasificación | Warnatz | París | Freiburg | EUROClass |
|---|---|---|---|---|
| Número de grupos | 3 | 3 | 3 | 5 |
| Utiliza células B memoria con y sin cambio de isotipo | Sí | No | Sí | Sí |
| Utiliza células CD21low | Sí | No | Sí | Sí |
| Utiliza células B transicionales | No | No | No | No |
| Utilidad como factor predictor | Esplenomegalia | Esplenomegalia y linfadenopatías | Esplenomegalia | Linfadenopatías, enfermedad granulomatosa y esplenomegalia |
| Población | Alemania | Francia | Alemania | Europa |
Conclusiones
Las células B de memoria, específicamente las que presentan cambio de isotipo, son fundamentales para las respuestas de anticuerpos dependientes de células T en el centro germinal y se ha encontrado que hasta 80 % de los pacientes con IDCV presentan defectos en el centro germinal y, por ende, en las células B de memoria, lo que ha demostrado que en este grupo de pacientes favorece el desarrollo de comorbilidades concomitantes como linfadenopatías, esplenomegalia, autoinmunidad y enfermedad granulomatosa, por lo cual se han realizado múltiples clasificaciones que utilizan en común a las células B de memoria para intentar realizar una clasificación de pacientes con IDCV y de establecer factores pronósticos.
De las clasificaciones con subpoblaciones de células B en IDCV, la EUROclass es la que hasta el momento ha demostrado mejor correlación entre la presencia de las comorbilidades y el pronóstico, sin embargo, no se encontró que alguno de los grupos tuviera mayor predicción de autoinmunidad y, por lo tanto, no puede usarse de forma aislada con tal propósito en los pacientes con IDCV.

