











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
En México, el TCE es la tercera causa de muerte que corresponde a muertes violentas y accidentes, con 35,567 defunciones y mortalidad de 38.8 por 100,000 habitantes.
En países tanto desarrollados como en vías de desarrollo, los vehículos a motor son la primera causa de TCE en personas jóvenes, particularmente. Las caídas son la causa principal de TCE en personas mayores de 65 años. En Estados Unidos, cada año se estima que 1.6 millones de personas sufren un TCE, de los que 800,000 aproximadamente reciben tratamiento ambulatorio y 270,000 requieren hospitalización. Cada año se registran 52,000 muertes y 80,000 personas con discapacidad neurológica secundaria a esta causa. En los países donde no hay guerra, la mayoría de TCE son causados por accidentes de vehículos de motor con 78% de los casos de TCE severo, de los cuales, 53% son accidentes por automóvil, 22% por accidentes de motocicleta y 3% por atropellamiento. Otras causas importantes son los accidentes laborales, con 19% de los casos. Las lesiones en eventos deportivos constituyen 1.8% de los casos y las agresiones representan 2%, pero se considera que menos de 10% de las agresiones son por arma de fuego. El TCE es la causa principal de muerte en pacientes menores de 40 años. Los accidentes de tránsito constituyen la causa más frecuente de trauma y tienen una alta tasa de mortalidad, que en Iberoamérica oscila entre 11 y 16 por 100,000 habitantes por año y constituyen la causa principal de TEC severo.1 En Iberoamérica, la incidencia de TEC es de 200 a 400 por cada 100,000 habitantes por año y es más frecuente en el sexo masculino, con una relación de 2:1 a 3:1, afectando a la población joven económicamente activa. Esto genera un costo tanto social como en la atención hospitalaria e incide en la economía y progreso de la sociedad. La mortalidad ronda 30% en los centros especializados en trauma.2
El TCE cursa con dos fases. La primera es debida al impacto directo: fractura, contusión, hemorragia puntiforme y subaracnoidea; y la segunda se debe a edema, hipoxia y hemorragia subsecuentes. El trauma directo produce contusión del parénquima cerebral y lesión axonal difusa en la sustancia blanca cerebral y del tronco encefálico. La lesión primaria desencadena una serie de alteraciones en el metabolismo cerebral que comprometen la hemodinámica intracraneal y la homeostasis iónica. El daño cerebral se acompaña de isquemia e hipoxia. En estas condiciones, la producción de energía va a depender de la glicólisis anaeróbica, que es un mecanismo poco eficiente de producción energética, condicionando la disminución de los niveles de ATP. Los niveles de ATP comienzan a bajar en forma sustancial en los primeros tres minutos de la hipoxia.3 La bomba sodio-potasio requiere una molécula de ATP para transportar dos iones de potasio al interior de la célula y tres iones de Na+ al exterior. La disminución de la producción de ATP conduce a la falla de la bomba Na-K, produciéndose la pérdida neta de potasio y ganancia de sodio intracelular que, debido al efecto osmótico, arrastra agua al interior de la célula. De igual forma, la bomba de calcio es dependiente de ATP y extrae dos iones de Ca++ por cada ATP hidrolizado, lo que conduce al aumento de la concentración de calcio intracelular.4
El daño primario es inmediato y no puede prevenirse o tratarse, ya que éste se ha completado antes de recibir atención médica. Si es grave, el paciente puede fallecer de manera simultánea. La mejor manera de mitigar el daño primario es la prevención con medidas como el uso del casco en motociclistas.5
Existen dos tipos de daño primario: el traumatismo craneal cerrado (TCC) y el traumatismo craneal penetrante (TCP). En el TCC, el impacto directo del cerebro contra el cráneo y el corte de las estructuras neurovasculares por las fuerzas de rotación o de rebote dan como resultado un daño en el cuerpo celular y los axones. Los accidentes de tráfico son colisiones a alta velocidad con una desaceleración muy rápida y son particularmente perjudiciales debido a que las estructuras neuronales, que residen en un compartimento lleno de líquido, se mueven durante la detención repentina del cuerpo en movimiento, chocando contra la bóveda craneal. Las estructuras se golpean tanto en el plano directo como en el opuesto del movimiento contra la lámina ósea interna. Ésta es la base del patrón de lesión por golpe-contragolpe donde se observa una lesión contusional en el cerebro profundo, que es el lugar del impacto del cráneo y 180 grados opuesto al lugar del impacto. Si hay fuerzas de rotación, las estructuras se tuercen y puede ocurrir desgarre. Ésta es la causa de la lesión axonal difusa y se observa comúnmente en TAC o MRI como hemorragias después del TCE.5 Un impacto traumático induce la respuesta inflamatoria sistémica, que induce edema cerebral y muerte neuronal.6 En el traumatismo craneoencefálico penetrante, la bóveda del cráneo es violada por un cuerpo extraño. El cuerpo invasor puede ser grande y moverse lentamente, como un cuchillo, o puede ser pequeño y en movimiento rápido, como una bala. En ambos casos, el cuerpo intruso lesiona las estructuras neuronales, vasculares y estromales a medida que atraviesa el cerebro. Si el objeto se mueve a una velocidad muy alta, el vacío creado por la estela del proyectil da lugar a la cavitación del tejido. Los proyectiles disparados pueden causar este tipo de lesión, dependiendo de la forma y la velocidad de entrada.
Las lesiones causadas por un TCE pueden clasificarse como focales o difusas. Las lesiones focales se producen en el lugar del impacto y los déficits neurológicos son atribuibles a estas áreas. Las áreas más propensas a recibir lesiones de este tipo son las lesiones orbitofrontales y en la región anterior del lóbulo temporal, ya que se encuentran sobre la superficie rugosa en la base del cráneo.7 La lesión difusa se circunscribe básicamente a la lesión axonal difusa (LAD) y a algunos casos de tumefacción cerebral difusa (swelling). Una LAD es el corte de los axones en la sustancia blanca cerebral, lo que causa la aparición déficits neurológicos no lateralizados, como la encefalopatía. Las consecuencias de este tipo de lesión pueden tener un retraso de aparición de hasta 12 horas después del trauma. La LAD se produce por efecto de fuerzas inerciales que actúan sobre los axones durante unos 50 ms en sentido lineal o angular (por ejemplo, en colisiones frontales), lo que produce la desconexión y ruptura de los axones (axotomía primaria). No obstante, la mayoría de los axones dañados (94%) son afectados por la axotomía diferida, que consiste en un aumento a la permeabilidad de Ca++ en los nodos de Ranvier que causa la destrucción celular. Ambas axotomías evolucionan desfavorablemente con cambios histopatológicos progresivos como la formación precoz de bulbos de retracción axonal, acumulación de microglia y presencia de tractos de degeneración walleriana, misma que puede identificarse como hemorragias petequiales en la materia blanca (especialmente subcortical) en la TAC y RMN después de un TCE; sin embargo, los resultados pueden aparecer sutiles o ausentes en las imágenes. Los pacientes que padecen una LAD están subreactivos desde el momento en que se inflige el traumatismo, porque la afectación axonal interrumpe las señales del sistema reticular activador ascendente y sus manifestaciones van desde una conmoción cerebral hasta la lesión axonal difusa grave.8
El swelling difuso puede presentarse tardía o precozmente asociado a otros tipos de lesiones focales (contusiones) y difusas (LAD) o como entidad única. Durante el desarrollo del daño secundario es posible encontrarlo con hipertensión intracraneal y otras lesiones anatómicas como la isquemia.5,8
El daño secundario. La lesión comienza rápidamente después de la fase primaria y puede continuar durante un periodo prolongado. La lesión cerebral secundaria es la causa principal de muerte hospitalaria tras un TCE; la mayoría son causadas por la inflamación del cerebro, con un aumento de la presión intracraneal (PIC) y la consiguiente disminución de la perfusión cerebral, que conduce a isquemia. Involucra disfunción y muerte de las neuronas, la glía y de estructuras de soporte. Se presume que la carga más importante de la lesión neurológica después de un TCE tiene que ver con esta lesión secundaria. Una amplia gama de mecanismos están implicados en la lesión secundaria e incluyen hipoxia, isquemia, radicales libres, aminoácidos excitatorios, desequilibrio de iones (como el calcio), la desregulación de temperatura y la inflamación. Esta respuesta cerebral también puede determinar cambios patológicos sistémicos como distrés respiratorio, diabetes insípida, síndrome de pérdida cerebral de sal o pirexia central. Horas después del TCE, el líquido que se acumula en el cerebro causa edema cerebral, aumenta la PIC y reduce el umbral de la presión arterial sistémica de la isquemia cerebral. Muchos intentos por desarrollar estrategias terapéuticas se han centrado en los procesos de lesión secundaria, pero, a pesar de los esfuerzos de investigación, el tratamiento clínico actual se limita principalmente a las medidas de apoyo con especial énfasis en el mantenimiento de la presión de perfusión y oxigenación de los tejidos (en especial el encéfalo), a minimizar la hipertensión intracraneal y al tratamiento del edema cerebral. La lesión secundaria es causada por ciertas condiciones intracraneales y extracraneales que disminuyen la oferta o aumentan el consumo de oxígeno, generando hipoxia cerebral tanto en el periodo inmediato al trauma como durante el manejo intrahospitalario. El aumento de la presión intracraneana por edema cerebral, hidrocefalia, lesiones ocupantes de espacio (hematoma epidural, subdural, hemorragia intracerebral) en presencia de hipotensión o normotensión condicionan la reducción de la presión de perfusión y del flujo sanguíneo cerebral. La hipoxia puede obedecer a causas extracraneales que reducen la oferta de oxígeno: obstrucción de la vía aérea por aspiración, trauma de tórax, hipotensión sistémica, anemia severa y depresión respiratoria de origen central.9
Varios agentes farmacológicos contra los radicales libres, los antagonistas del N-metil-D-aspartato y los bloqueadores de los canales de calcio han sido investigados en un intento por evitar la lesión secundaria asociada a una lesión cerebral traumática, pero ninguno ha demostrado ser eficaz.5,7
La hipoxia y la hipoperfusión son reconocidas como los factores principales que contribuyen a la lesión cerebral secundaria. El daño cerebral es más susceptible a estados hipóxico-isquémicos debido a los estados de alteración de la autorregulación vascular cerebral. Las áreas más susceptibles son el hipocampo y las regiones distales de la corteza. La fiebre, los estados sépticos y las crisis comiciales aumentan el metabolismo cerebral, motivo por el que los efectos de la isquemia serían, teóricamente, aún más devastadores. Se han asociado resultados desastrosos en pacientes con TCE grave que presentan un episodio de hipotensión (con presión sistólica por debajo de 90 mmHg). El daño microvascular difuso se asocia a pérdida de la autorregulación vascular cerebral y a la pérdida de integridad de la barrera hematoencefálica. La laceración de la microvasculatura exacerba esta lesión. El daño microvascular contribuye al edema vasogénico que se observa después de un TCE. La hiponatremia, a menudo asociada por diferentes mecanismos al TCE, es un factor determinante de mal pronóstico, dado que propicia edema intracelular.5
Es necesario entender la patogénesis multifactorial del TCE, donde varias vías celulares inician la respuesta primaria y secundaria al daño. El cerebro está considerado un sitio inmunológicamente privilegiado, como evidencia de lo contrario se encuentra la fisiopatología del TCE. La migración de leucocitos a través de la disrupción de la barrera hematoencefálica resulta en la activación de las células residentes del SNC, como microglia y astrocitos. La infiltración de células inmunes periféricas y la activación de células residentes generan subsecuentemente la producción intratecal de citocinas, marcadores importantes de neuroinflamación. Las citocinas pueden propiciar neurotoxicidad, aumentando la excitabilidad y propagando la respuesta inflamatoria.10
La isquemia postraumática activa una cascada de eventos metabólicos que culminan en la generación de especies reactivas de oxígeno (ERO), aminoácidos excitatorios (comúnmente glutamato y aspartato), citocinas y agentes proinflamatorios. La activación de receptores NMDA y AMPA causan exitotoxicidad celular por la entrada masiva de calcio a las células ocasionando neurodegeneración. Algunos factores tanto isquémicos como no isquémicos ocasionan la liberación de ERO de la mitocondria. Las ERO producen daño neurodegenerativo a membranas celulares, a proteínas intracelulares y a ácidos nucleicos por mecanismos de peroxidación; además, estimulan la activación de las fosfolipasas A2 y C, que hidrolizan los fosfolípidos de membrana, liberando ácido araquidónico. Se ha observado en laboratorio que un aumento en la síntesis de ácidos grasos libres, leucotrienos y tromboxanos se asocia a un mal resultado. Se ha notado un incremento en la producción de citocinas proinflamatorias tales como IL-1, IL-6, TNF-α, que, se presume, son generadas por activación de la microglia. Estas citocinas inducen una respuesta celular inflamatoria exuberante que se supone responsable de astrogliosis, edema y la destrucción del tejido.11
El TCE ocasiona un aumento del potasio intracelular, altera el potencial de la membrana y de los mecanismos reguladores de la Na+/K+ATPasa, predisponiendo a la célula a despolarizar. El potasio también aumenta el consumo de oxígeno por los astrocitos, lo que priva a las neuronas de dicho gas. El TCE grave provoca una reducción del Mg++ extracelular, lo que se refleja en glicólisis anormal, la respiración celular alterada, la fosforilación oxidativa disminuida (lo que aumenta la generación de ERO) y la biosíntesis de ADN, ARN y proteínas.12,13
Al restablecerse el flujo sanguíneo cerebral, se produce la lesión por reperfusión. En situaciones de isquemia/reperfusión, la enzima xantina deshidrogenasa se tranforma en xantino oxidasa.14 En condiciones de isquemia, el aumento del consumo de ATP tiene como consecuencia la acumulación de catabolitos purínicos xantina e hipoxantina, los cuales utilizan el oxígeno como sustrato. En el momento de la reperfusión y al aumentar la disponibilidad de oxígeno, estos catabolitos son metabolizados por la xantina oxidasa, convirtiéndolos en superóxido y éste, por acción de la superóxido dismutasa en presencia de hierro (Fe+2,3) se transforma en peróxido de hidrógeno. La enzima NADPH oxidasa y el ciclo de óxido-reducción del hierro constituyen otras fuentes de producción de especies reactivas del oxígeno durante la reperfusión, aunque su papel es menor que el de la xantina deshidrogenasa. Los radicales libres destruyen la membrana celular mediante la peroxidación lipídica. Por otro lado, alteran la función de algunas enzimas como la sintetasa de glutamina, transformando el glutamato en glutamina. Existe evidencia de que la lesión producida por las especies reactivas del oxígeno alcanzan su actividad máxima entre 20 y 24 horas posteriores al daño, lapso en el que la intervención terapéutica podría reducir la magnitud del daño cerebral secundario.15
El traumatismo craneoencefálico se clasifica como leve, moderado o grave con base en el grado de consciencia o la escala de coma de Glasgow (ECG). En el TCE leve o contusión (ECG 13-15), los pacientes han experimentado pérdida de la consciencia menor de 30 minutos y las quejas que se presentan incluyen dolor de cabeza, confusión y amnesia. Hay una recuperación neurológica completa, a pesar de que algunos de estos pacientes tienen dificultades de concentración o memoria pasajeras.16 En el TCE moderado (ECG 9-13), el paciente se encuentra letárgico o estuporoso.
Clínicamente, los pacientes con TCE moderado requieren hospitalización y pueden necesitar una intervención neuroquirúrgica; además, están asociados a más probabilidades de hallazgos anormales en las técnicas de neuroimagen. Estos pacientes también pueden desarrollar el síndrome postconmoción. El síndrome postconmoción se refiere a un estado de inestabilidad nerviosa después de un TCE leve o moderado. Las características principales son fatiga, mareo, pupila anisocórica, signos de daño cerebral grave.16 En el TCE grave o severo (ECG 3-8), el paciente se encuentra en un estado comatoso, no puede abrir los ojos, seguir órdenes y sufre de lesiones neurológicas significativas. Por lo general, tiene una neuroimagen anormal; es decir, en la tomografía computarizada (TAC/TC) se observa fractura del cráneo o hemorragia intracraneal. Estos pacientes requieren ingreso a la unidad de cuidados intensivos (UCI) y la toma de medidas urgentes para el control de la vía aérea, ventilación mecánica, evaluación o intervención neuroquirúrgica y monitorización de la presión intracraneal (PIC). La recuperación es prolongada y generalmente incompleta. Un porcentaje significativo de pacientes con TCE grave no sobrevive más de un año. Una lesión en la cabeza durante el periodo de recuperación puede resultar en síndrome del segundo impacto, que se observa sobre todo en niños y adolescentes. Se ha asociado significativamente con resultados clínicos peores.16
La baja en el flujo sanguíneo cerebral (FSC) propicia un círculo vicioso que ocasiona un aumento en la PIC y disminución del FSC. El cerebro no puede almacenar sustratos y tiene una alta demanda de oxígeno, por lo tanto, requiere un aporte circulatorio continuo. Esta condición se deteriora en más de 50% de los pacientes dentro de las primeras 24 horas y suele mantenerse por cinco días. El flujo sanguíneo cerebral (FSC) en condiciones normales se satisface con 15% del gasto cardiaco. El FSC es relativamente constante, a pesar del cambio en la presión arterial media (PAM). Es necesario que la presión de perfusión cerebral (PPC, equivalente a PAM-PIC) se mantenga cerca de 60 mmHg. Si es menor, habrá isquemia, y, si es mayor, aumentará el volumen sanguíneo intracraneal.7,5 Fisiológicamente, el SNC tiene la capacidad de amortiguar los cambios en la PIC (rango normal = 10 ± 5 mmHg) como la obliteración de las cisternas y ventrículos mediante la evacuación de líquido cefalorraquídeo (LCR) y la expulsión de hasta 7% del volumen sanguíneo intracraneal fuera del lecho venoso cerebral. Cuando la capacidad de regulación se agota, el paciente experimenta un aumento exponencial de su PIC, lo cual ocurrirá de forma progresiva o de forma periódica. En este último caso, habrá ascensos de 40-80 mmHg con una duración de cinco a 20 minutos. El aumento de la PIC reducirá la PPC, por lo que inevitablemente se incrementará la isquemia cerebral preexistente y, al no ser ésta homogénea (por la compartimentalización de las meninges), la mayor magnitud de presión se localizará en las áreas donde exista una masa postraumática, que irá disipándose mediante el desplazamiento del tejido cerebral, llegando al enclavamiento.5
El hematoma epidural se forma entre la Tabla interna del cráneo y la duramadre, como consecuencia de la ruptura de la arteria y/o la vena meníngea media, y en la fosa posterior, por ruptura de los senos transversos y sigmoideos. Se localiza con mayor frecuencia en la región temporal o temporoparietal, predominando en el lado derecho. Entre 1 y 3% de los casos el hematoma es bilateral. La incidencia reportada es de 2.5 a 4% y es más frecuente entre la segunda y tercera década de la vida. La causa más frecuente son los accidentes de vehículo automotor (50%), seguida de caída accidental (30%) y traumatismo con objeto contundente (10%). De forma característica, en 50% de los casos se acompaña de pérdida inicial de la consciencia, seguida de un periodo de lucidez mental y deterioro rápidamente progresivo del estado neurológico. El coma sin periodo de lucidez mental puede ser la única forma de presentación entre 20 y 50% de los pacientes. La anisocoria se manifiesta en cerca de 50% de los casos, especialmente cuando la forma de presentación es el coma. La presencia de lesiones supratentoriales con efecto de masa provocan el desplazamiento y herniación ipsilateral del uncus del hipocampo (localizado en la cara interna del lóbulo temporal) y del gyrus parahipocampal sobre la tienda del cerebelo o tentorio y produce compresión del nervio motor ocular común, cuya manifestación clínica es la midriasis. Si no se evacúa el hematoma expansivo, se producirá compresión y descenso de las amígdalas cerebelosas y del tronco encefálico a través del foramen magnum, conduciendo a la muerte del paciente. La anisocoria se asocia a un pronóstico variable determinado por la relación entre el lado del HE y el de la midriasis. Cuando la midriasis es ipsilateral al HE, es reversible y se asocia a un curso evolutivo benigno, siempre que la evacuación del HE se lleve a cabo en los primeros 70 minutos. En estos casos, hay compresión y elongación de las fibras pupilares parasimpáticas del nervio motor ocular común a nivel del hiato tentorial, que revierte al evacuar el hematoma. La presencia de midriasis contralateral o bilateral es indicador de mal pronóstico (mortalidad de 74%), debido a que implica daño mesencefálico severo e irreversible con afectación de las vías pupilares centrales.3,5,17
El hematoma subdural es más frecuente que el epidural, se presenta entre 20 y 30% de los TEC severos. La causa de HS difiere con la edad del paciente. Los accidentes en vehículo automotor constituyen la causa más común entre los pacientes de 20 a 40 años de edad (56%), y las caídas accidentales en los mayores de 65 años (56%). El hematoma subdural aparece como consecuencia de la ruptura de venas puente entre la corteza cerebral y los senos venosos. La colección subdural se acumula entre la duramadre y la aracnoides y, dado que esta última no se adhiere al cráneo en los sitios de sutura, el hematoma se distribuye a lo largo de la superficie cerebral, proporcionándole el aspecto tomográfico de concavidad interna. Los factores que se asocian a mal pronóstico y determinan la evolución del paciente son la desviación de la línea media, hematoma subdural con un grosor > 18 mm, la presencia de contusión cerebral y lesiones extracraneanas que con frecuencia acompañan al hematoma subdural. La desviación de la línea media < 10 mm o la presencia de HS con un grosor < 10 mm se asocia a 85-90% de supervivencia. La diferencia negativa (desviación de la línea media menor que el grosor del hematoma) menor de -5 mm tiene buen pronóstico y se asocia a una supervivencia de 85%.3,17
La HSA se presenta entre 33 y 61% de los casos de TEC severo y es detectable precozmente en la TAC inicial. Las caídas accidentales constituyen la causa más común (47%), seguida de los accidentes en vehículo automotor (34%). Afecta con mayor frecuencia a pacientes del sexo masculino (70%) entre los 30 y 50 años de edad. La HSA se localiza comúnmente en la convexidad de los hemisferios cerebrales (67%) y en las cisternas basales (40%), y en la mayor parte de los casos están asociadas a contusión cerebral (72%), hematoma subdural (20-40%), obliteración de cisternas mesencefálicas (50%) y desviación de la línea media (34%). La presencia de HSA eleva de 2 a 3.6 veces el riesgo de evolución desfavorable y muerte en el paciente con TEC severo. La existencia de lesión intracraneana con criterio neuroquirúrgico (efecto de masa y desviación de la línea media > 5 mm) no evacuada en las primeras tres horas y la compresión de cisternas basales se asocian a mal pronóstico. El riesgo de evolución desfavorable aumenta con la severidad de los hallazgos tomográficos. Existe correlación entre el puntaje en la escala de coma de Glasgow al ingreso y los hallazgos tomográficos basados en la escala propuesta por Greene. Los pacientes con 6 a 8 puntos en la ECG y con TAC grado I tienen 38% de probabilidad de buena recuperación en la evaluación de el electroencefalograma, mientras que aquéllos con TAC grado III tienen 14% de probabilidad de buena evolución. La presencia de TAC grado IV se asocia a discapacidad severa en 82% de los casos. Cuando el puntaje en la ECG es de tres a cinco, la presencia de hallazgos de grado I en la TAC, se asocia a 70% y de grado IV a 92% de probabilidad de discapacidad severa, respectivamente.18
Los hematomas intraparenquimatosos son consecuencia del movimiento brusco del encéfalo en el interior del cráneo, que provoca contusión cerebral y ruptura de vasos sanguíneos. Generalmente, se localizan en los lóbulos frontal (43%) y temporal (24%, de éstos 50% en cara lateral, 35% en el área polar y 15% en cara inferior). El hematoma intraparenquimatoso puede producir efecto de masa. Con frecuencia no es visible en la tomografía inicial, sino después de 24 horas y hasta 10 días posteriores al trauma. El pronóstico se determina por factores clínicos (edad, ECG postreanimación, alteración de los reflejos del tallo encefálico), tomográficos (fractura de bóveda craneana, presencia de lesiones intracraneanas asociadas, magnitud y localización de las lesiones, volumen de los hematomas, compresión de cisternas mesencefálicas y severidad del edema), nivel de PIC y retrasos en la detección de la lesión e intervención neuroquirúrgica. Los hematomas localizados en los ganglios basales tienen mal pronóstico (mortalidad de 60%).17,19
El trauma craneoencefálico está asociado a un aumento en los niveles séricos de catecolaminas. Las catecolaminas son responsables de los depósitos de neutrófilos, mientras que los corticosteroides causan la salida de los neutrófilos de la circulación. Las catecolaminas aumentan la cuenta leucocitaria, introduciendo las células marginadas al pool circulante.20 La respuesta de fase aguda también se caracteriza por leucocitosis al ingreso, por lo que es probable que la cuenta de células blancas sirva como indicador adicional al diagnóstico y pronóstico del trauma de cráneo.21,22
La barrera hematoencefálica se abre al mismo tiempo que el trauma y se cierra 60 minutos después del daño. Después del trauma, la microglia se vuelve hipertrófica, la cual se expresa en antígeno mayor de histocompatibilidad I y II, que se presenta en los linfocitos y ocasiona la activación a nivel sistémico de los mismos.20,23
Otro mecanismo por el cual los leucocitos están asociados al daño cerebral es la ruptura traumática de microvasos, seguida de la oclusión de la misma. Los leucocitos son menos deformables que los eritrocitos y se requiere un gradiente de presión mayor para forzarlos a pasar por los capilares de diámetro pequeño. En condiciones de baja presión de perfusión, los capilares deben comportarse como un colador y atrapar los leucocitos para incrementar la cuenta leucocitaria. Tras el atrapamiento, los leucocitos forman un área común en contacto con el endotelio y no serán desalojados hasta que la perfusión retorne a la normalidad. La oclusión mecánica en los capilares puede volverse más evidente como resultado del aumento del número de sustancias citotóxicas, que incrementan la interacción con el endotelio.
Muchos estudios se han usado para predecir el desenlace y el pronóstico de los pacientes con TCE severo. La escala de coma de Glasgow es uno de los mejores indicadores, la cual es complementada con variables como la edad, reactividad pupilar y hallazgos tomográficos que han sido probados como buenas variables pronósticas. Rara vez se utilizan parámetros directamente relacionados con daño cerebral, como hiperglucemia y leucocitosis, los cuales están estrechamente vinculados con el pronóstico.22
Los pacientes con trauma de cráneo severo tienen una cuenta leucocitaria significativamente alta que corresponde a la severidad del trauma. Se ha encontrado una relación entre la cuenta leucocitaria y el puntaje de la escala de coma de Glasgow, la respuesta pupilar y la presencia de hemorragia subaracnoidea.21 En un grupo de pacientes, la cuenta leucocitaria era mayor en quienes tenían peor pronóstico.21,22 La leucocitosis (> 12,000 103/µl) es el pronóstico marcado como más significativo de los días de estancia hospitalaria.24,25
Existen marcadores bioquímicos que ayudan a predecir la severidad del problema, en este caso, la cuenta leucocitaria puede ayudar a medir la respuesta inflamatoria sistémica del paciente y su respuesta adecuada. El cerebro está privilegiado inmunológicamente y muestra un reclutamiento leucocitario atípico secundario a un daño, lo que cambia la idea de la relación entre el cerebro y la periferia.26,27 Los leucocitos migran al sistema nervioso central después de algunos tipos de daño como el TCE, siendo éstos los principales mediadores de la respuesta secundaria tras el daño. En comparación con otros tejidos donde los leucocitos son rápidamente reclutados tras el daño, hay un momento en el que el reclutamiento de leucocitos es sometido a un retraso, lo cual provee una ventana donde se inhibe el daño subsecuente mediado por leucocitos.12,23,26
La leucocitosis está comúnmente asociada a causas infecciosas o inflamatorias, pero puede ocurrir en variedad de situaciones. En muchos estudios, la leucocitosis está asociada a mortalidad y morbilidad. Se ha estudiado particularmente la relación de la leucocitosis con morbilidad y mortalidad en eventos vasculares y cerebrales. Combinada con otros modelos, puede contribuir a predecir el riesgo de mortalidad del paciente26 y de severidad del trauma en pacientes inestables que no pueden ser trasladados o cuando no se disponga de estudios de imagen de forma inmediata.27,28
Existen pocos estudios que muestren la correlación entre leucocitosis y lesión intracraneal en pacientes con TCE atendidos en los servicios de urgencias, por lo que sería un marcador pronóstico de gran utilidad en pacientes atendidos.29,30
Material y métodos
Tipo de estudio: Observacional.
Diseño de estudio: Estudio de cohorte, prospectivo longitudinal.
Población de estudio: Se incluyeron pacientes atendidos en el área de urgencias con TCE. Al ingreso se registraron signos vitales, se tomó una muestra para biometría hemática y se recopilaron estudios de imagen. Posteriormente, se tomaron mediciones de leucocitos a las 24 horas del ingreso. Pacientes mayores de 18 años y menores de 35 que ingresaron con diagnóstico de TCE.
Se elaboró formato de consentimiento informado y firmado, el cual se presentó a los familiares del paciente.
Criterios de selección
Criterios de inclusión: Mayores de 18 y menores de 35 años de edad con traumatismo craneoencefálico.
Criterios de no inclusión: Pacientes con herida penetrante en cráneo. Pacientes menores de 18 años. Pacientes que no cuenten con BH al ingreso. Pacientes con más de seis horas de evolución.
Criterios de eliminación: Pacientes que decidan no participar en el estudio. Circunstancias que no permitan determinar alguna de nuestras variables de estudio. Pacientes que no cuentan con información completa para el protocolo.
Tamaño de muestra
Nivel de confianza 95%, poder de la prueba 80%, relación de pacientes con lesión y sin lesión 1:1.
Frecuencia esperada de leucocitosis en pacientes con lesión 60%.
Frecuencia esperada de leucocitosis en pacientes sin lesión 30%.
Riesgo relativo de detectar de 2%.
El tamaño mínimo de muestra es de 96 sujetos.
Se calculó conforme a la fórmula de χ2 para proporciones en sistema STACALC.
Análisis estadístico
Análisis univariado con proporciones simples y relativas de las variables dicotómicas y de las continuas medidas de tendencia central y de dispersión.
Análisis bivariado de riesgo relativo, intervalos de confianza 95%, χ2, valor de p, t de Student para muestras pareadas y t de Student para muestras independientes.
Análisis multivariado: se realizará a través de regresión logística múltiple para buscar el mejor modelo que explique la relación entre leucocitosis y el tipo de lesión y otras terceras variables.
Metodología operacional
Se realizó estudio observacional, prospectivo, longitudinal y multicéntrico. Se incluyeron pacientes atendidos en el área de urgencias con TCE, mayores de 18 años y menores de 35. Al ingreso se tomó registro de signos vitales, se recolectó muestra para biometría hemática y se recopilaron los estudios de imagen disponibles. Posteriormente, se tomaron mediciones iguales a las 24 horas del ingreso.
Se elaboró una hoja de consentimiento informado y firmado, la cual se presentó a los familiares del paciente.
Se usaron dos grupos para comparar las constantes vitales, laboratorio y estudios de imagen en grupo I, TCE leve; y grupo II, TCE severo. Nivel de confianza 95%, poder de la prueba 80%, relación de pacientes con lesión y sin lesión 1:1. Frecuencia esperada de leucocitosis en pacientes con lesión 60%. Frecuencia esperada de leucocitosis en pacientes sin lesión 30%. Riesgo relativo de detectar de 2%.
Resultados
Se reunieron 96 pacientes entre 18 y 35 años, siendo la media de 27 años, resultando una población de 78 hombres y 18 mujeres. De estos, se encontraron con lesión intracraneal 51 hombres (65.4%), 10 mujeres (55.6%), siendo un total de 61 pacientes, y 35 sin lesión intracraneal (36.5%). No se detectó significancia estadística entre sexo y lesión intracraneal (p = 0.435). De los 61 pacientes con lesión intracraneal, hubo una incidencia de 15 (15.6%) pacientes con hemorragia subaracnoidea (HSD), 16 (16.7%) con hematoma subdural, 16 (16.7%) con contusión hemorrágica y 14 (14.6) con hematoma epidural.
Por grupos de edades: se detectó presencia de lesión intracraneal en un total de 24 pacientes menores de 25 años y 37 mayores de 25 años, siendo 63.2% y 63.8%, respectivamente. Sin significancia estadística por grupo de edades, p = 0.950. Por tipo de lesión: en menores de 25 años, cuatro pacientes presentaron hemorragia subaracnoidea (HSA) (10.5%), cinco pacientes hematoma subdural (13.2%), nueve pacientes contusión hemorrágica (23.7%) y seis pacientes hematoma epidural (15.8%). En pacientes mayores de 25 años, se observó una frecuencia de 11 pacientes con hemorragia subaracnoidea (19%), 11 con hematoma subdural (19%), siete con contusiones hemorrágicas (12.1%) y ocho con hematoma epidural (13.8%).
Se observó también que de los pacientes que ingresaron con diagnóstico de TCE, 54 refirieron ingesta de alcohol y 30 presentaron lesión intracraneal (55.6%).
De los 96 pacientes incluidos en el estudio con una edad media de 28 años, en pacientes con lesión intracraneal se observó a su ingreso una media de conteo total de leucocitos de 16,396.07 en pacientes con lesión y a las 24 horas un total de leucocitos de 13,246 en pacientes con lesión intracraneal (Figura 1).
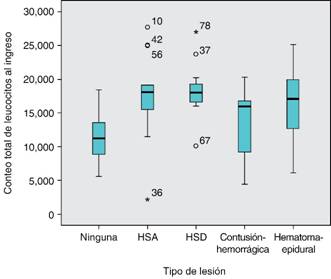
Figura 1: Muestra los valores de leucocitos al ingreso divididos por tipo de lesión en pacientes atendidos con TCE (p < 0.0001).
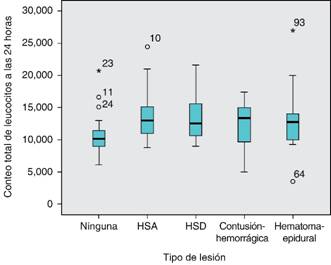
De los pacientes atendidos con hemorragia subaracnoidea (HSA) (15), se detectó a su ingreso una media de leucocitos de 17,718 103/μl y de 13,970 103/μl a las 24 horas del trauma, p = 0.000 y 0.001, respectivamente, lo que refiere una diferencia estadísticamente significativa. En pacientes con hematoma subdural (HSD) (16) se encontró a su ingreso una media de leucocitos de 18,212 10^3/μl y de 13,319 10^3/μl a las 24 horas, con una p = 0.000 y 0.003, respectivamente, lo cual refiere una diferencia estadísticamente significativa. En pacientes con contusión hemorrágica (16) se observó a su ingreso una media de leucocitos de 13,225 10^3/μl y de 12,501 10^3/μl a las 24 horas, una p = 0.091 y 0.027, respectivamente, también con diferencia estadísticamente significativa. En pacientes con hematoma epidural (HE) (14) se detectó a su ingreso una media de leucocitos de 16,527 10^3/μl y de 13,240 10^3/μl a las 24 horas con una p = 0.000 y 0.019, respectivamente, con una diferencia estadísticamente significativa.
La leucocitosis estuvo presente en 44 pacientes (45.8%) a su ingreso con una proporción mayor en los casos de lesión intracraneal (n = 43, 97.7%), respecto a los casos sin lesión (n = 1, 2.3%) con una diferencia estadísticamente significativa (p < 0.0001). La leucocitosis mostró una proporción notoriamente menor a las 24 horas sólo en ocho pacientes (8.3%), un poco más alta en los casos con lesión intracraneal (n = 6, 75.0%) respecto a los casos sin lesión (n = 2, 25.0%), estadísticamente similares (p = 0.482) (Figura 2).
Discusión
Existen marcadores bioquímicos que ayudan a predecir la severidad del problema; en este caso, la cuenta leucocitaria puede ayudar a medir la respuesta inflamatoria sistémica del paciente y su respuesta adecuada.
El cerebro está privilegiado inmunológicamente y muestra un reclutamiento leucocitario atípico secundario a un daño, lo que cambia la idea de la relación entre el cerebro y la periferia. Los leucocitos migran al sistema nervioso central después de algunos tipos de daño como el TCE, siendo éstos los principales mediadores de la respuesta secundaria tras el daño. En comparación con otros tejidos donde los leucocitos son rápidamente reclutados tras el daño, hay un momento en el que el reclutamiento de leucocitos es sometido a un retraso, lo cual provee una ventana donde se inhibe el daño subsecuente mediado por leucocitos.
Existen pocos estudios que muestren la correlación entre leucocitosis y lesión intracraneal en pacientes con TCE, por lo que sería un marcador pronóstico de gran de utilidad en pacientes atendidos.
Conclusión
La leucocitosis temprana (primeras seis horas postrauma) es un factor de mal pronóstico y predictivo de lesión cerebral en pacientes con trauma craneoencefálico.
Se observó que, en los pacientes con TCE, la leucocitosis estuvo presente en los casos con lesión intracraneal evidente en estudio de imagen con una diferencia significativa de quienes no la tuvieron, por lo que puede establecerse como un factor pronóstico.

