











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Las vasculitis sistémicas son un grupo de enfermedades de difícil diagnóstico cuyo pronóstico siempre es incierto. El complejo del SPR se define como la combinación de hemorragia alveolar difusa acompañada de glomerulonefritis rápidamente progresiva.1 Existen varios mecanismos, entre los principales se describen la actividad de anticuerpos anticitoplasma de neutrófilos (ANCA), anticuerpos antimembrana basal glomerular (anti-MBG), así como la microangiopatía trombótica.1,4
La hemorragia alveolar difusa a su vez se caracteriza por la tríada clásica de hemoptisis, infiltrados radiológicos difusos y caída brusca del hematocrito, ya que se ha demostrado que la patología pulmonar subyacente es una vasculitis de pequeños vasos que implica arteriolas, vénulas y capilares alveolares.1,3
La asociación a patología renal es una forma de glomerulonefritis proliferativa y focal acompañada de necrosis fibrinoide, la cual es causada por daño de los capilares y las membranas basales con fuga de los eritrocitos, seguido de una afluencia de macrófagos, fibrinógeno y la formación de la proliferación de células extracapilares (medias lunas).1,2
Las tres causas más comunes de presentación SPR son las vasculitis ANCA + de vasos pequeños asociados a (anti-MBG) la enfermedad de Goodpasture (GP) y el lupus eritematoso sistémico (LES).1,5
Las vasculitis ANCA + describen cuatro principales síndromes sistémicos: la granulomatosis con poliangeítis antes granulomatosis de Wegener, la poliangeítis microscópica, el síndrome de Churg-Strauss (CSS) y el síndrome pulmón riñón idiopático cuyas características clínicas y patológicas muestran diferencias sutiles. Las vasculitis ANCA-negativos, tales como la púrpura de Henoch-Schönlein y la nefropatía IgA son de presentación más rara.1,2,4
Caso clínico
Paciente masculino de 63 años de edad portador de diabetes mellitus tipo 2 e hipertensión arterial sistémica de 10 años de diagnóstico, ingresa al servicio de urgencias por cuadro clínico de dos meses de evolución caracterizado por pérdida de peso de 8 kg de forma no intencionada, malestar general, tos con expectoración hemoptoica, hipertermia no cuantificada y datos de dificultad respiratoria. Ingresa a urgencias en malas condiciones generales, fiebre de 38 oC y datos clínicos de dificultad respiratoria, sus paraclínicos iniciales: Hb 9.5 g/dL, Hto 27.1%, leucocitos 11,000, neutrófilos 96%, Linf 5%, Eos 1.5%, QS: Cr 2.1 mg/dL, Urea 74 mg/dL, BUN 37 mg/dL, el resto de estudios paraclínicos sin alteraciones, radiografía de tórax con presencia de infiltrados bilaterales y broncograma aéreo, gasometría arterial: pH 7.34, PO2 62, PCO2 47, HCO3 18.5, EB -1.8, lactato 2.8 manejado con hidratación y antibioticoterapia. Es ingresado al servicio de medicina interna con diagnóstico de neumonía atípica, dentro del protocolo de estudio se solicitó PCR para Tb, marcadores tumorales y cultivos resultando negativos, se continuó manejo antimicrobiano con levofloxacino; sin embargo, persistió con deterioro respiratorio y presentó de forma súbita hemoptisis masiva e insuficiencia respiratoria aguda que requirió manejo avanzado de vía aérea, se solicitó estudio radiográfico que evidenció infiltrado bilateral difuso (Figura 1), estudio gasométrico con hipoxemia y acidosis respiratoria pH 7.11, PO2 45, PCO2 65, HCO3 12.1, EB -8.7, Sat 83%. Se solicitó su ingreso a UCI donde persistió con falla respiratoria y renal, paraclínicos Hb 5.8 g/dL, leucocitos 12,780, Ntf 95%, Linf 1.9%, Cr de 3.5 mg, BUN 90 mg/dL, urea 202 mg/dL. Se solicitó estudio tomográfico con evidencia de múltiples áreas de consolidación e infiltrados bilaterales, así como broncograma aéreo (Figura 2). Se aplicó manejo con medidas de protección alveolar, ventilación en posición prono durante 24 horas y sustitución de la función renal con hemodiálisis (HD). Ante sospecha de etiología autoinmune se inició manejo con metilprednisolona (1 g c/24 horas) durante cinco días con mejoría parcial, sin remisión total de la sintomatología. Se solicitó perfil inmunológico reportando: VSG corregida 12 mm/h, Ac. antinucleares ANA (+) 1:80, factor reumatoide negativo, C3 y C4 normales, IgG 806, IgM 10, mg/dL, IgE 341 mg/dL. Perfil de ANCAS por inmunoensayo: C-ANCA/PR3 (+) con nivel 1.35 ng/dL, P-ANCA/MPO (-), Ac. antimembrana basal glomerular (-). La biopsia renal reveló: glomerulonefritis por complejos inmunes con patrón proliferativo extracapilar mixto (activo y fibroso), proliferación endocapilar segmentaria acompañada de lesiones esclerosantes. La inmunohistoquímica: IgG, IgM (+) con patrón granular focal, fibrinógeno (+) en algunos segmentos del mesangio, en semilunas y zonas de necrosis fibrinoide (Figuras 3, 4 y 5). Albúmina con patrón lineal por hiperfiltración en las membranas basales glomerulares y en las membranas basales tubulares ante hallazgos histopatológicos y bioquímicos así como baja respuesta a manejo con esteroide. Se agregó al manejo ciclofosfamida a dosis de 500 mg/m2/SC sin obtener respuesta, continuando con hipoxemia refractaria acidosis metabólica persistente. Hipotensión sin respuesta a apoyo vasopresor y falla renal ocasionando la muerte.
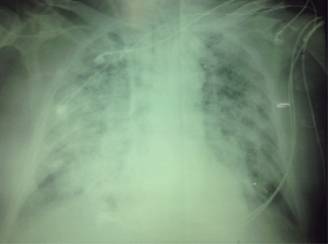
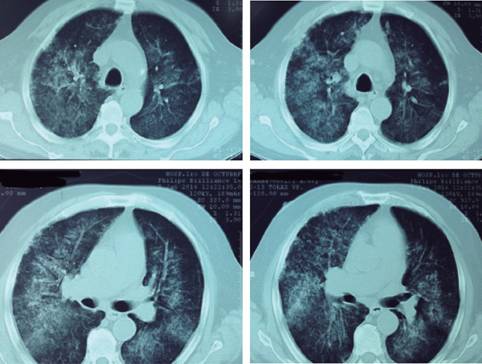

Figura 3: Microfotografía 40x teñida con PAS en la que se observa una lesión activa (semiluna celular) con necrosis fibrinoide.
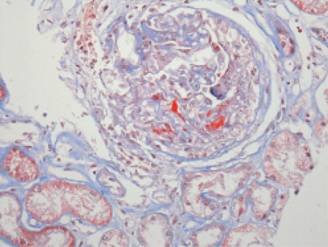
Figura 4: Microfotografía 40x teñida con metenamina en la que se observa una lesión activa (semiluna celular) con necrosis fibrinoide.
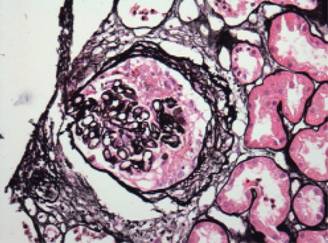
Figura 5: Microfotografía 40x teñida con tricómico de Masson en la que se observa una lesión activa (semiluna celular) con necrosis fibrinoide.
Fisiopatología
La patología primaria en la mayoría de los SPR incluye la inflamación y necrosis de los vasos sanguíneos de pequeño calibre, con consecuente daño alveolar e inflamación glomerular a través de la infiltración de neutrófilos en el endotelio vascular.4
A nivel pulmonar existe una degradación de la pared capilar y la matriz intersticial que afecta arteriolas, capilares y vénulas trayendo como consecuencia la destrucción y necrosis de los vasos sanguíneos, todo esto es mediado por complejos inmunes que dan lugar a una capilaritis necrótica, lo que se traduce en extravasación de eritrocitos al interior del alvéolo manifestándose como hemorragia alveolar.4,10
A nivel renal se presenta una glomerulonefritis proliferativa focal, necrosis fibrinoide y trombos microvasculares, con múltiples variaciones patológicas como necrosis fibrinoide, microangiopatía in situ y granulomatosis necrotizante. Existe formación de media luna, infiltración intersticial, fibrosis y atrofia tubular, que son factores de mal pronóstico.3,10
Los anticuerpos anticitoplasma de neutrófilos (ANCA) son anticuerpos antineutrófilos dirigidos contra la célula, existen numerosos componentes de ANCA dirigidos contra los componentes de los neutrófilos, pero sólo dos formas de ANCA son específicas para la proteincinasa 3 (PR3) y mieloperoxidasa (MPO), éstas están presentes en los gránulos azurófilos de los neutrófilos y pueden expresarse en la superficie de los polimorfonucleares.4,11
El papel patogénico de los ANCA (MPO y PR3) en el citoplasma de los neutrófilos no estimulados se inicia por citoquinas (TNF alfa e IL-8) que se expresan en la superficie celular generando una reacción de estos antígenos con los ANCA, este proceso conduce a la activación de los granulocitos y la liberación de moléculas de adhesión de leucocitos con células endoteliales vasculares, finalizando en una necrosis celular y apoptosis del endotelio vascular.2,4
La unión de ANCA a las membranas de neutrófilos activa la célula que conduce a la liberación de enzimas líticas, IL-8 y radicales libres de oxígeno. Los neutrófilos se agregan posteriormente en el endotelio causando inflamación y daño a las microvasculatura.11
Anatomía patológica
La hemorragia alveolar difusa (HAD) tiene tres tipos histológicos: capilaritis pulmonar, hemorragia alveolar blanda y daño alveolar difuso.5
La capilaritis se caracteriza por eritrocitos y/o hemosiderina intersticial, necrosis fibrinoide de las paredes capilares y trombos de fibrina en capilares septales así como neutrófilos en el intersticio y en espacios alveolares adyacentes con coágulos de fibrina en septos.3
La hemorragia alveolar blanda se caracteriza por la presencia de sangre en los espacios alveolares sin inflamación ni destrucción de los capilares, vénulas y arteriolas del intersticio pulmonar.3,11
El daño alveolar difuso se caracteriza por edema de los septos alveolares con mínima inflación, congestión capilar septal alveolar, microtrombosis de vasos pequeños y formación de membranas hialinas en espacios alveolares.3,11
Las lesiones histológicas renales en vasculitis ANCA positivos se caracterizan por GN focal o difusa, con áreas de necrosis fibrinoide segmentaria.3
La proliferación extracapilar con formación de semilunas acompaña a la afectación glomerular casi de forma constante.3
Clasificación
Se clasifican de acuerdo con los mecanismos patogénicos:
Vasculitis sistémicas ANCA positivos: (60-70% de los casos). Granulomatosis con poliangeítis (GW), poliangeítis microscópica (PAM), síndrome de Churg-Strauss (SCHS) y SPR-ANCA positivos idiopáticos.
Enfermedad por Ac-AMBG: síndrome de Goodpasture.
Enfermedades autoinmunes: LES es la que tiene la asociación más fuerte al SPR, la esclerosis sistémica, la artritis reumatoide, la enfermedad mixta del tejido conectivo y la polimiositis.
Vasculitis sistémicas ANCA negativos: púrpura de Henoch-Schönlein, crioglobulinemia mixta y la enfermedad de Behcet.
Vasculitis ANCA positivos por drogas: propiltiouracilo, metimazol, hidralazina, alopurinol, d-penicilamina, sulfasalazina y fenitoína.
Epidemiología
El SPR tiene una incidencia de 10 casos/millón habitantes/año, se presenta predominantemente en poblaciones caucásicas entre 40 y 55 años de edad, en más de 70% de los casos existe una asociación significativa entre las ocupaciones agrícolas y la exposición a disolventes. Sucede con mayor frecuencia en el género masculino, como factores de riesgo se menciona la presencia de tabaquismo, patologías reumatológicas como artritis reumatoide (AR) y LES.4
El síndrome de Churg-Strauss es una vasculitis sistémica y pulmonar rara, con una incidencia de menos de tres casos/millón, con mayor incidencia en pacientes asmáticos (64 casos/millón), es más frecuente en el género masculino 2:1, con inicio a edad mediana, se presentan pANCA en 30 a 50% de los casos.4
Enfermedad anti-MBG (Goodpasture) es la causa más rara del SPR con una incidencia anual de un caso/millón, los hombres son cuatro veces más propensos, se manifiesta entre 20 y 30 años de edad, muestra un segundo pico en las mujeres mayores de 60 años, se asocia a un alto riesgo de glomerulonefritis debido a la fuerte asociación con el HLA DR15 y DR4.4
La hemorragia alveolar como complicación de la nefritis lúpica es rara y siempre tiene un pronóstico grave con cifras de mortalidad entre 30 y 50%.4
Cuadro clínico
El cuadro clínico inicial se caracteriza por síntomas respiratorios que rápidamente progresan, disnea de reposo, fiebre y tos, en algunos casos la sintomatología inicial incluye astenia, adinamia y pérdida de peso. Cuando la enfermedad tiene una evolución más lenta, la hemoptisis se manifiesta de forma súbita en aproximadamente 70% de los casos acompañada de un descenso en las cifras de hemoglobina4 y, en la mayoría de los casos, de la necesidad de ventilación mecánica, puede mostrar datos inespecíficos como lesiones purpúreas en piel, mialgias, artralgias y polineuropatía. Se presentará siempre un deterioro súbito de la función renal que varía de días a semanas.5,6
Diagnóstico
Los hallazgos de laboratorio pueden ser inespecíficos al inicio, entre ellos el más frecuente es un descenso agudo de la hemoglobina, con detección de anemia normocítica, normocrómica, puede o no existir alteración de las demás líneas celulares. La elevación brusca de azoados es característica, se comporta como una lesión renal aguda, con reporte de sedimento urinario activo (proteinuria incluso en rangos de síndrome nefrótico o bien datos de síndrome nefrítico como hematuria, cilindros hemáticos o dismorfia eritrocitaria) que son datos indicativos de glomerulonefritis activa, con rápida progresión de la falla renal.1,4,11
La lesión renal asociada a un sedimento urinario activo es de vital importancia para el diagnóstico, la proteinuria y la hematuria son comunes, cuando ambas están presentes es indicativo de daño de la membrana glomerular debido a glomerulonefritis.4
En cuanto a estudios de imagen la radiografía de tórax o tomografía computarizada puede revelar una distribución de infiltrados de sombreado difuso, tendiendo hacia las zonas más bajas a la consolidación franca que imitan la apariencia de un síndrome de insuficiencia respiratoria aguda.4
La detección de anticuerpos séricos como anticuerpos anti-MBG, ANA y/o ANCA es clave para el diagnóstico, cabe destacar que el nivel de título de ANCA no se considera parte de los criterios diagnósticos de vasculitis sistémica, la presencia de un ANCA positivo dirigido contra el antígeno PR3 se correlaciona con glomerulonefritis subyacente en 85% de los casos, pANCA dirigido contra MPO tiene reactividad cruzada con múltiples condiciones inflamatorias y autoinmunes, por lo que la interpretación clínica es de suma importancia al momento del diagnóstico, los anticuerpos anti-MBG detectados por inmunoensayo tienen una sensibilidad de 95 a 100% y una especificidad de 90 a 100% en el diagnóstico de SGP.1,9,13
La broncoscopia con lavado bronquioalveolar se considera el estudio de elección para evidencia de HAD en pacientes con infiltrado bilateral y difuso sin evidencia clara de hemoptisis, en el que se realiza la detección microscópica de hemosiderófagos.3,7
La biopsia renal y pulmonar provee el diagnóstico definitivo, en la microscopia de luz hay una GNF con formación de media luna en el compartimiento de la cápsula de Bowman (proliferación extracapilar) en más de 50% de los glomérulos.9,14
Con la técnica de inmunohistoquímica hay un patrón de deposición, ya sea capilar, mesangial, granular o lineal a lo largo de la membrana basal glomerular y difiere dependiendo de la etiología. En el síndrome de Goodpasture existen depósitos lineales a lo largo de la membrana basal glomerular, mientras que en una GNF ANCA se desencadena la formación de depósitos inmunes (GNF pauciinmune) y en vasculitis mediada por complejos inmunes es posible observar una imagen con la deposición granular de IgG, IgM, IgA o complemento.4,12
El diagnóstico diferencial del síndrome pulmón riñón se clasifica de la siguiente forma:
Clasificación de Chapel Hill Jennette para Vasculitis ANCA +
Diagnóstico diferencial
Síndrome antifosfolípido con vasculitis y/o embolia pulmonar.
Enfermedades del tejido conectivo mixto (esclerosis sistémica, polimiositis).
Púrpura trombocitopénica trombótica.
Enfermedades infecciosas que implican el riñón y el pulmón (hantavirus, citomegalovirus, Legionella, mycoplasma, leptospira, tuberculosis).
Por otra parte, una enfermedad renal primaria puede conducir a una enfermedad pulmonar e imitar la imagen de un síndrome pulmón riñón:
Insuficiencia renal aguda con edema pulmonar y hemoptisis urémica.
Tromboembolismo en el síndrome nefrótico: trombosis de la vena renal y/o pulmonar embolia.
Inmunosupresión en la enfermedad renal y una neumonía.
Por el contrario, una enfermedad pulmonar primaria puede conducir a la enfermedad renal e imitar la imagen del síndrome pulmón riñón:
La infección de vías respiratorias con insuficiencia prerrenal renal y/o glomerulonefritis postinfecciosa o hematuria en la nefropatía IgA
El cáncer de pulmón con nefritis por complejos inmunes.7
Tratamiento
El éxito en cuidados intensivos (UCI) del manejo de pacientes con SPR depende de un rápido reconocimiento, exclusión de la infección y tratamiento agresivo específico desde el inicio de la sintomatología.4,6
La mayoría de los pacientes son ingresados a UCI con diagnóstico de sospecha o confirmado de SPR, este último es por lo regular secundario a un deterioro agudo de la función respiratoria y renal, presencia de hemorragia alveolar difusa e insuficiencia respiratoria o bien por infección severa agregada y estado de inmunosupresión secundario a terapia biológica. Cabe señalar que en reportes de caso se ha descrito que hasta 75% de los pacientes fallecen por sepsis, ya que el riesgo de infección nosocomial es extremadamente alto.4,8
Debido a la hemorragia alveolar difusa en la lesión pulmonar, grandes volúmenes corrientes o cambios de presión pueden exacerbar el daño en la microvasculatura pulmonar, por lo que desde el ingreso del paciente debe implementarse la ventilación de protección pulmonar, tal como se utiliza en el tratamiento del SIRA con volúmenes corrientes de 6-8 mL/kg de peso predicho y la presión meseta inferior a 30 cm de H2O.
Medidas generales
El manejo del estado de choque, ya sea por hipovolemia debido a la pérdida aguda de volumen sanguíneo o bien por la respuesta inflamatoria sistémica, debe tratarse de manera oportuna con reanimación hídrica y hemoderivados, y de considerarse necesario, con aporte de vasopresor.
La terapia de reemplazo renal temprano está indicada en pacientes con lesión renal aguda, considerando un nivel BUN de 72 mg/dL, no hay que esperar a que el paciente evolucione a un estadio III de AKIN o presente criterios absolutos de sustitución renal.10,15
Manejo específico
Durante muchos años se utilizó el esteroide como monoterapia; sin embargo, se observó que los resultados a un año no eran favorables, con una alta tasa de mortalidad de hasta 20-30%.8
La introducción de la ciclofosfamida en combinación con esteroides anunció una alternativa en el tratamiento de las vasculitis, pues se apreció una disminución de 50% en la mortalidad a los cinco años en comparación con el tratamiento con glucocorticoides como monoterapia; no obstante, el uso de ciclofosfamida por sí solo se considera factor independiente de riesgo de desarrollar cáncer de vejiga.8,11
La ciclofosfamida es un agente alquilante que produce la apoptosis celular a través de la síntesis de ADN y ARN, sus efectos secundarios dependen de la dosis acumulada, predispone a la cistitis hemorrágica y mielosupresión crónica, aumenta el riesgo de trastornos linfoproliferativos y toxicidad gonadal.4,6,8
La remisión se logra más comúnmente con pulsos de metilprednisolona (0.5 a 1 g/día) durante 3-5 días, continuando con un régimen diario de prednisona oral a 1 mg/kg/día por tres meses, se recomienda asociar ciclofosfamida (15 mg/kg) administrada cada 2-3 semanas con un régimen oral diario (1-2 mg/kg/día).8,13
Otros agentes biológicos utilizados para inducir la remisión incluyen el metotrexato y micofenolato, contraindicados en enfermedad renal grave, además se ha demostrado la superioridad de la ciclofosfamida en el mantenimiento de la función renal normal a los seis meses de diagnóstico, el uso de rituximab para remisión de cuadro no ha probado ser superior a la ciclofosfamida.4,8
El método de mantenimiento ha sido objeto de estudio, actualmente continúa siendo muy variable el tipo de inmunosupresor y la duración del tratamiento, los glucocorticoides a dosis bajas durante un mínimo de 18 meses junto con un agente economizador de esteroides es bien aceptado, aunque puede extenderse hasta dos años en pacientes con PR3 ANCA positivos ante mayor riesgo de recaída. Actualmente hay tendencia al cambio de ciclofosfamida por azatioprina con duración de 3-6 meses desde el diagnóstico.4,8
La terapia de mantenimiento con metotrexato, leflunomida y micofenolato no han mostrado ninguna ventaja sobre la azatioprina.4,13
Las terapias biológicas utilizadas para el mantenimiento incluyen el TNF-α, infliximab, etanercept y rituximab, los cuales se han asociado a complicaciones infecciosas y han demostrado ser tan eficaces como la ciclofosfamida.4,8
Los regímenes de inducción con ciclofosfamida son eficaces en 70 a 90% de los pacientes, pero se han asociado a altas tasas y eventos de mortalidad, el tratamiento con rituximab ha conducido a tasas de remisión de 80 a 90% entre los pacientes con vasculitis asociada a ANCA refractaria y puede ser más seguro que los regímenes de ciclofosfamida; no obstante, las tasas de remisión fueron altas con ambos regímenes, la ingesta de rituximab no se asoció a la reducción de eventos adversos, pero ambos se asociaron a una mortalidad 18%.16
Plasmaféresis
En presencia de la enfermedad renal grave se ha detectado beneficio a corto plazo con el uso de plasmaféresis en términos de recuperación de la función renal en los primeros 18 meses.10
El estudio MEPEX en 2007 reveló que el uso de plasmaféresis reduce el riesgo de desarrollar insuficiencia renal crónica a los tres meses, sin impactar de forma significativa en la mortalidad, en pacientes con Cr < 5.6 mg/dL.17
Aún existe controversia sobre el beneficio a largo plazo del uso de plasmaféresis. Cabe destacar que el mecanismo de acción no está bien establecido, se han encontrado menores títulos de ANCA posterior a plasmaféresis, se presume que elimina gran parte de las citoquinas proinflamatorias, el sistema del complemento y factores de coagulación. Es probable que disminuya la progresión de la enfermedad renal en pacientes que desarrollan insuficiencia renal grave, aunque no ha probado beneficio en la supervivencia a largo plazo y no existe evidencia de su papel en el tratamiento de enfermedades menos graves.1,8,10
Pronóstico
La edad y la creatinina sérica en la presentación «de inicio» siguen siendo los más fuertes predictores de la supervivencia.
En múltiples patologías se ha observado que el retraso en el tratamiento se asocia a un aumento en la mortalidad.5
El uso de plasmaféresis de forma temprana en pacientes con afección renal disminuye la necesidad de terapia de reemplazo renal de forma permanente, sin encontrarse hasta el momento datos que apoyen la disminución de mortalidad. En este año se lleva a cabo el estudio PEXIVAS que tiene como objetivo aleatorizar pacientes con la finalidad de demostrar las repercusiones en la mortalidad del uso oportuno de plasmaféresis.
Conclusión
El manejo del SPR requiere un equipo multidisciplinario, el ingreso a unidad de cuidados intensivos siempre está justificado. El papel del intensivista es tratar el síndrome de insuficiencia respiratoria aguda y el inicio de terapia de sustitución renal de forma temprana con la finalidad de limitar las secuelas respiratorias y evitar la progresión a falla renal crónica, pero sin dejar atrás el manejo del choque séptico que es la principal complicación al iniciar terapia inmunosupresora. Es importante señalar que según la revisión bibliográfica existen pocas alternativas terapéuticas que modifiquen el pronóstico, por lo que de forma obligada debe iniciarse a la brevedad terapia con esteroide sistémico y manejo inmunosupresor.
En el caso particular de nuestro paciente, de acuerdo con hallazgos de laboratorio y biopsia de riñón así como evolución clínica se concluyó en síndrome pulmón riñón idiopático. El paciente se ingresó con más de 48 horas de retraso en el tratamiento ventilatorio, fue sometido a manejo de SIRA con medidas de protección alveolar y ventilación en posición PRONO para mejorar la oxigenación rebelde a otras medidas, obteniendo importante mejoría en los niveles de oxigenación y ventilación; sin embargo, con dificultad para alcanzar niveles de anticuerpos antimembrana basal así como biopsia renal. Se inició de manera oportuna esteroide sistémico en bolos y sustitución de la función renal; no obstante, de manera tardía el manejo inmunosupresor y pese al soporte multiorgánico el paciente evolucionó de forma catastrófica y finalmente falleció, lo cual asociamos al retraso en tratamiento, aunque debe recalcarse que hasta la fecha lo único que disminuye la mortalidad en este tipo de pacientes es la asociación de esteroide sistémico y terapia inmunosupresora de manera oportuna.

