











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
In a 2017 report issued by the International Committee on Taxonomy of Viruses (ICTV), papillomaviruses (PVs) were classified into two subfamilies, Firstpapillomavirinae and Secondpapillomavirinae, both of which belong to the Papillomaviridae family.1 Papillomaviruses are non-enveloped, double-stranded DNA viruses, having an icosahedral symmetry that consists of a circular genome, approximately 8 kilobase (kb) in length.2
Papillomaviruses that infect cattle are included in the Firstpapillomavirinae subfamily. To date, a variety of different papillomavirus types (n = 25) from five genera (Delta-, Dyoxi-, Dyokappa-, Epsilon-, and Xipapillomavirus) have been identified from clinical specimens obtained from cutaneous or mucosal epithelial lesions from infected cattle.3
Bovine PVs (BPV) are immunosuppressive infectious agents causing tumoral lesions that when occurring on the udder and teat can lead to economic losses in dairy cattle. Therefore, effective preventive strategies should determine the dominant BPV type present through its genome characterization.
In this study, the full genome of the BPV obtained from wart material taken from dairy cattle udders in Turkey was characterized. The result of our study confirm that the dominant type of BPV-1 reported herein and in previous studies in Turkey have high homology with other worldwide BPV-1 species.
Materials and methods
A sample from a wart lesion was obtained from a non-pregnant Holstein heifer belonging to a Turkish dairy herd (from Bursa Province) where five other cows had widespread warts similar to papilloma on other parts of the body (Figure 1). Milking was performed by machine and the owner had reported loss of milk production.

Figure 1 Papilloma lesions on the mammary gland and udder of a dairy heifer from a daire infected with Papillomavirus.
Viral DNA was extracted from the homogenized wart tissue with a PureLink Viral RNA/DNA (Invitrogen, Carlsbad, CA, USA) extraction kit. The polymerase chain reaction (PCR) method was performed using Phusion high-fidelity DNA polymerase (Invitrogen, Carlsbad, CA, USA). Two overlapping 4.1-kb sections of the papilloma virus genome were amplified by using primer sequences described by Regnard et al.4
Using a primer walking approach, the entire genome of a PV was cleaned up and sequenced. For primer walking, custom primers were designed (Primer3 software) to be complementary to the ends of the known sequence to perform consecutive elongation steps (Table 1). The repeated cycles of sequencing reactions and primer design were continued until all contigs were joined and the whole sequence was obtained. Annealing temperature of primers was calculated as 52 °C. PCR products were purified with the ExoSAP-IT kit (USB Europe GmBH, Staufen, Germany) and sequenced with an ABI 3730XL automated sequencer (Applied Biosystems, Foster City, CA), using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems).
Table 1 Papillomavirus Primer sets used for amplification of viral DNA.
| Primer name | Primer sequences | Deltapapillomavirus 4 isolates* complete genome |
Our primer base length |
|---|---|---|---|
| TCO 1-2_F2 | 5’- TAGCCAGGATGAGGATTTTGTT-3’ | 989-1010 | 22 |
| TCO 1-2_R2 | 5’- ACCGAGGCCTGCTCTGAT-3’ | 3360-3343 | 18 |
| TCO 1-2_F3 | 5’- ATATGCTTTGGCTGCAGGAT-3’ | 1850-1869 | 20 |
| TCO 1-2_-R1 | 5’- CACTGGCTATTGGCTGTGTTT-3’ | 4025-4045 | 21 |
| TCO A-B_F2 | 5’- GGACAGAAGTGGGACCACAG-3’ | 5120-5139 | 20 |
| TCO A-B_R2 | 5’- ACTGCAAATGCTGGACAGG-3’ | 7239-7221 | 19 |
| TCO A-B_F3 | 5’- CACCACCCAAACAACAGATG-3’ | 6004-6023 | 20 |
* The primers targeted regions in the accession numbers MG977494 and MG263871 from GenBank database.
Phylogenetic analysis was performed using the complete BPV genome sequences available from the GenBank database. A tree was generated by the Neighbor-Joining (NJ) method based on the Tamura-Nei model, using MEGA Software (version 6.0).5
We used RDP46 and MEGAX7 to investigate potential polymorphism and recombination in alignment of four BPV-1 reference strains and our complete BPV sequence (Table 2). Tajima’s Neutrality Test was processed as implemented in MEGA. We have also applied the codon-based test of neutrality between mentioned sequences which is implemented in MEGA. This analysis was conducted based on Nei-Gojobori Algorithm Method. In recombination investigation, an automatic, RDP, and three manual methods (BOOTSCAN, GENECOV, MAXCHI) were selected. Pairwise calculation was estimated with Clustal W by using two matrix softwares SDT and MatGAT.6,7
Results and discussion
Clear sequence reads produced a 7946-nucleotide (nt)-long consensus contig. The sequence data were compared using the GenBank8 database, and eight open reading frames (early genes E1, E2, E4, E5, E6, and E7 and the late genes L1 and L2 of PVs) were identified. The genome sequence of TR-BPV-type 1 has been deposited in GenBank under accession number MH197482 (Figure 2).
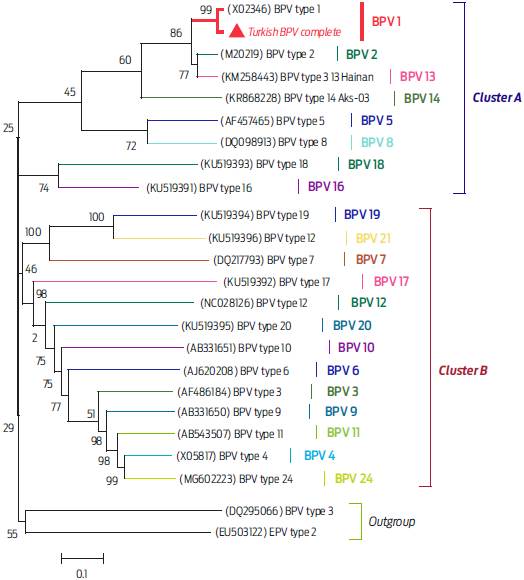
Figure 2 Phylogenetic tree for genetic relationships of BPVs and Turkish BPV type 1 strain. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Tamura-Nei method5 and are in the units of the number of base substitutions per site. The analysis involved 24 nucleotide sequences. Codon positions included were 1st+2nd+3rd+Noncoding. All positions containing gaps and missing data were eliminated. BPV subtypes on tree were showed at taxa with green marked branches. Turkish BPV sequence (MH197482) marked by a red triangle. Two main clusters, including different BPV types, have been defined after phylogenetic analysis (Cluster A and B). CPV and EPV were determined as members of outgroup.
To clarify the genetic relationships between Turkish BPV type 1 and other BPV subtypes, a phylogenetic tree was constructed (Figure 2). The analysis based on the NJ5 method showed that the Turkish BPV strain was genetically related to the BPV type 1 isolate (accession number X02346) and located in a separate branch (Figure 2).
Tajima’s Neutrality Test was used to determine selection hotspots. Observed average pairwise diversity (π) has been calculated 0.011342. Some SNPs were observed in nucleotide alignment; however, these polymorphisms have not led to critical changes at codon translation level. Negative Tajima’s D value indicates homogeneity and non-unity of SNPs in Table 2a. Additionally, no unique recombination has been detected among reference strains and our sequence in RDP. Results of the performed tests indicated that the E2 gene region was more stable than other genes of BPV, resulting in a lesser density of SNPs in the gene (Table 2b).
Table 2.a Tajima’s Neutrality Test.
| m | S | p s | Θ | π | D |
|---|---|---|---|---|---|
| 5 | 61 | 0.024361 | 0.011693 | 0.011342 | -0.227377 |
Abbreviations: m = number of sequences, n = total number of sites, S = Number of segregating sites, p s = S/n, Θ = p s/a1, π = nucleotide diversity, and D is the Tajima test statistic
This analysis involved 5 amino acid sequences. The coding data was translated assuming a genetic code table.
All positions containing gaps and missing data were eliminated (complete deletion option).
There were a total of 2504 positions in the final dataset.
Table 2.b Codon-based Test of Neutrality for analysis between sequences.
| BPV-1 sequences | Std. error | ||||
|---|---|---|---|---|---|
| KY746722 PP002 Morocco | 1.108 | 0.6239 | -0.209 | 2.3547 | |
| MF045489 GZLZ China | 0.2701 | 1.0707 | 0.1235 | 1.9859 | |
| AB626705 Japan:Hokkaido | 0.5339 | 0.2865 | -0.061 | 0.7507 | |
| X02346 BPV | 0.8348 | 0.9019 | 0.9514 | 0.6561 | |
| MH197482 Turkish BPV | 0.0202 | 0.0493 | 0.4543 | 0.5130 | |
| p-value | |||||
The probability of rejecting the null hypothesis of strict-neutrality (dN = dS) (below diagonal) is shown. Values of P less than 0.05 are considered significant at the 5% level and are highlighted. The test statistic (dN - dS) is shown below the diagonal.
dS and dN are the numbers of synonymous and nonsynonymous substitutions per site, respectively. The variance of the difference was computed using the analytical method. Analyses were conducted using the Nei-Gojobori method. This analysis involved 5 nucleotide sequences. All positions containing gaps and missing data were eliminated (complete deletion option). There were a total of 2504 positions in the final dataset.
Full genome comparison results between Turkish BPV type 1 and other BPV type 1 strains obtained from the GenBank database showed that our sequence shared an identity greater than 99% with the sequence AB626705 from Japan, and 51.3-99.6% identity rates with other complete PV genome sequences, which include different hosts obtained from the GenBank database (Figure 3 and 4).9,10
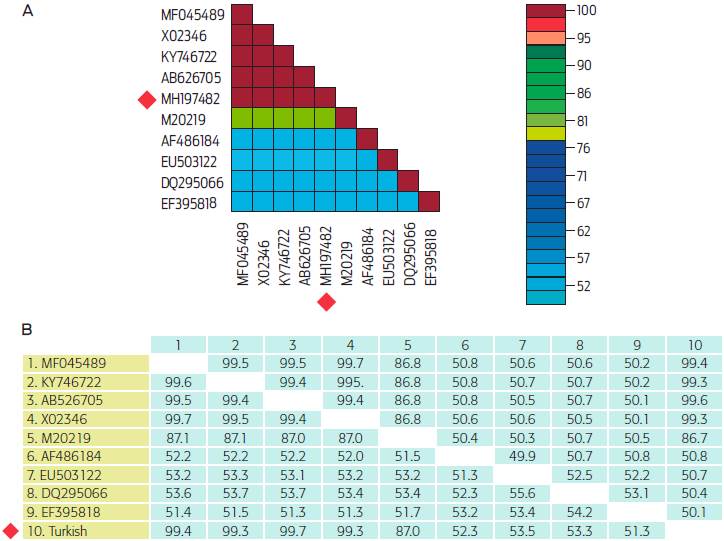
Figure 3 Pairwise similarity rates between reference sequences and the sequence belonging to this study. (A) Result of SDT analysis. (B) Result of MatGAT analysis. Both analyses involved 10 nucleotide sequences. Sequences and their accession numbers; BPV 1 GZLZ from China (MF045489), BPV 1 (X02346); BPV 1 PP02 from Morocco (KY746722); BPV 1 from Japan Hokkaido (AB626705); BPV 2 (M20219); BPV 3 (AF486184); EPV 2 (EU503122); CPV 3 (DQ295066); SPV 1 (EF395818); Turkish BPV (MH197482) marked by a red square.

Figure 4 Amino acid changes in alignment of BPV-1 sequences. The E2 gene is more stable than other genes, whereas the most variable region was detected as L2 gene region for translation assessment of our sequence.
PV infections are known to be associated with the formation of benign skin tumors, known as papillomas, warts, and fibropapillomas in cattle.11 It is known that BPV infection is common in Turkey12,13 and leads to economic losses in the dairy cattle industry.
The knowledge of the entire genome of BPV-1, which, unlike many other PVs, can cause infection in other species, will be informative for the phylogenetic analysis of this virus in different species. The BPV 1 genome is divided into three main parts: the long control region (LCR), essential for replication and transcription, the early region open reading frames (ORFs) (E1 to E8 genes), and the late region ORFs for the major L1 and the minor L2 capsid proteins.14 The genome sequence of the Turkish BPV strain was closely related with other PV complete genomes of bovine origin published in the GenBank database (Figure 2). Amino acid comparisons of the Turkish BPV strain in this study and other PVs were found to be similar in sequence results. The nucleic acid identity of the TR-BPV type 1 strain was found to be most closely related to Chinese, Japanese, and Moroccan BPV type 1 strains, between 99.3% and 99.6% (Figure 3).
In general, two primer pairs have been used in phylogenetic analysis studies that deal with characterization and/or typing of BPVs.13,15-17 One of these consists of degenerate primers FAP59/FAP64 that target conserved sequences of HPV capsid protein gene L1, which differentiate BPVs, as well as a variety of PVs.18 A second primer pair, namely MY09/MY11,19 was designed also from a conserved region of the HPV L1 gene, and is widely used for identifying PVs in humans and a wide range of animals. Villiers et al.20 indicate that identity ratios should be greater than 98-99% (1-2% sequence diversity) for variant determination, and that ratios between 90% and 98% (2-10% sequence divergence) can be determined as subtypes based on the L1 gene sequence identities of PVs. However, if the sequence identity ratio is below 90%, then the sequence belongs to a different type.16 When our complete genome analysis was compared with other PV full genome sequences circulating around the world, our BPV strain showed an almost 99.4% sequence homology with other BPVs. In this context, it could be named as a BPV type 1 genomic variant.
In a previous study in Turkey,13 detection, type classification, and molecular characterization of BPVs, were performed using FAP 59-64 and MY 09-11 primers. Among a total of 36 samples, 30 reacted with FAP primers and 22 with MY primers. Data obtained with these primers targeting a partial L1 gene region gave different results for the same samples (while FAP primers detected 9 samples, MY primers classified 16 samples as BPV type 1), and FAP primers were deemed deficient for the diagnosis of BPV infection. In addition, using two different primer pairs has a second disadvantage for the characterization of BPV types. In the phylogenetic analysis of the mentioned study, the same strains were amplified with FAP and MY primer pairs and located in different branches as different types. Consequently, these partial primer pairs were not able to give a consensus result and conflicted in discriminating of the virus type. To avoid these disadvantages, we chose to analyze the complete genome of the field BPV strain in our study. Also, it is known that multiple types of PVs are frequently present in the same lesion,21 which is a problem in characterization studies. The other reason for choosing full genome analysis for this research was to avoid missing these multiple infections and to elucidate the genomic structure of the dominant types circulating previously claimed by two reports in the country.12,13Eventually, this data will serve not only to elucidate the genetic structure, but will also contribute to prepare synthetic peptide vaccines for the control of this infection.
As for SNPs densities, our study showed that L1 has been the most conserved gene region, as emphasized in a previous report.22 The most variable gene has been determined as non-coding region (NCR). High consensus has been observed on amino acid motifs between BPV-1 sequences (Figures 3 and 4). The Turkish BPV1 has especially exhibited similar motifs with the reference strain Japan: Hokkaido. This also confirmed that there have been no unique amino acids changes in terms of mutation evaluation.
This is the first characterization of a full-length genome of BPV type 1 in Turkey. To understand the pathogenicity and diversity of BPV infection in our country, detailed genomic characterization studies are necessary. Furthermore, we suggest that studies are conducted to characterize the complete genome analyses of the different BPV subtypes obtained from cattle in Turkey.

