










Servicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkVeterinaria México OA
versión On-line ISSN 2448-6760
Veterinaria México OA vol.1 no.1 Ciudad de México jul./sep. 2014
https://doi.org/10.21753/vmoa.1.1.331
Artículos científicos
Cambios genéticos en los genes internos del virus de influenza porcina aislado en México
aDepartamento de Medicina y Zootecnia de Cerdos, Facultad de Medicina Veterinaria y Zootecnia, Universidad Nacional Autónoma de México, Av. Universidad 3000, Col. Cd. Universitaria, Del. Coyoacán, CP 04510, Distrito Federal, México.
bLaboratorio de Microbiología e Inmunología, Unidad de Investigación Multidisciplinaria, Facultad de Estudios Superiores Cuautitlán, Universidad Nacional Autónoma de México, Campo Cuatro, Km. 2.5 Carretera Cuautitlán-Teoloyucan, San Sebastián Xhala, Cuautitlán Izcalli, Estado de México, CP. 54714.
cInstituto de Biotecnología, Universidad Nacional Autónoma de México, Avenida Universidad 2001, Col. Chamilpa, Cuernavaca, Morelos, 62210.
Hasta el momento, en México sólo se ha publicado un reporte en el que se detallan los cambios genéticos y la divergencia evolutiva en los llamados genes internos de la polimerasa básica 1 (PB1), polimerasa básica 2 (PB2), polimerasa ácida (PA), nucleoproteína (NP), proteína de matriz (M) y la proteína no estructural (NS) del virus de influenza porcina que se extendió durante la pandemia entre 2009 y 2010. El objetivo de este estudio fue evaluar la divergencia evolutiva y los cambios genéticos de las cepas de virus de influenza porcina aisladas en México entre los años 2009-2010. Para caracterizar la historia evolutiva y la relación filogenética entre las cepas de virus aisladas, se utilizó una prueba de probabilidad de máxima verosimilitud usando un bootstrap filogenético con 1000 réplicas. La relación filogenética entre los genes PB1, PB2, PA, NP M y NS corresponde a aislamientos de virus porcinos similares a los consignados en otros países. Sin embargo, el gen PA del virus A/swine/Mexico/Qro35/2010 (H1N1) está estrechamente relacionado a un subtipo viral H3N2 humano. Los genes PB2, NP y M de los virus de influenza porcina mexicanos que proliferaron durante 2009 y 2010, configuran su distribución de acuerdo con los genes internos triplemente recombinantes (TRIG). En nuestro estudio se demuestra la presencia de un gen de PA de un virus humano H3N2 en la cepa de virus porcino A/swine/ Mexico/Qro35/2010 (H1N1).
Palabras clave: Virus de influenza porcina; Caracterización filogenética; Genes internos; México
To date, only one report has been published in Mexico detailing the genetic changes and evolutionary divergence in the so-called internal genes basic polymerase-one (PB1), basic polymerase-two (PB2), polymerase acid (PA), nucleoprotein (NP), matrix protein (M), and non-structural protein (NS) of the swine influenza virus that circulated during the 2009 pandemic and in 2010. The aim of this study was to evaluate the evolutionary divergence and genetic changes in these internal genes of the swine influenza virus strains isolated in Mexico in 2009-2010. To characterize the evolutionary history and the phylogenetic relationship among the isolated virus strains, a Maximum Likelihood Model Test analysis and a phylogenetic bootstrap test with 1000 replicates were performed. The phylogenetic relationships among the PB1, PB2, PA, NP, M and NS genes correspond to similar swine virus isolates reported in other countries. However, the PA gene from the A/swine/Mexico/Qro35/2010 (H1N1) virus is closely related to a H3N2-subtype human virus. The PB2, NP, and M genes of the Mexican swine influenza viruses that circulated during 2009 and 2010 maintained the distribution of the Triple Reassortant Internal Genes (TRIG). Significantly, our study demonstrates the presence of a human H3N2 virus PA gene in the A/swine/Mexico/Qro35/2010 (H1N1) virus.
Keywords: Swine influenza virus; Phylogenetic characterization; Internal genes; Mexico
Introducción
El genoma del virus de influenza A se caracteriza por poseer una sola cadena de ARN segmentado en 8 piezas. Estos segmentos codifican 12 proteínas: la polimerasa básica 2 (PB2), codificada por el segmento 1: polimerasa básica 1 (PB1), PB1-F2, y N40, codificadas por el segmento 2; la polimerasa ácida (PA), codificada por el segmento 3; la hemaglutinina (HA), codificada por el segmento 4; la nucleo-proteína (NP), codificada por el segmento 5; la neuraminidasa (NA), codificada por el segmento 6; la proteína de matriz M1 y el canal de iones M2, ambas codificadas por el segmento 7, y finalmente dos proteínas no estructurales (NS1 y NS2) que codifica el segmento 8. Hasta ahora se han identificado 17 variantes de HA (Tong et al., 2013) y 9 de NA , que proveen las bases para la clasificación de los tipos de influenza A en subtipos virales (Vincent et al., 2008).
Los virus de influenza se encuentran entre los patógenos humanos más importantes, debido a que causan epidemias estacionales y pandemias de manera ocasional. Del siglo pasado, en 1918, 1957 y 1968, se recuerdan estas tres principales pandemias de influenza. En marzo y principios de abril del 2009, los organismos de salud detectaron el primer caso de influenza A/H1N1pdm 09 en México y Estados Unidos. Debido a la facilidad de transmisión entre los humanos, el virus se diseminó rápidamente alrededor del mundo y causó la primera pandemia de influenza del siglo XXI (Dawood et al., 2009). La caracterización genética y los análisis filogenéticos mostraron que la cepa pandémica fue de origen porcino. Los investigadores encontraron que los genes PB1, PB2, PA, HA, NP y NS derivaban de un aislamiento estadounidense, de un virus porcino triple recombinante, mientras que los genes NA y M provienen de un virus porcino de linaje euroasiático (Garten et al., 2009).
También documentaron, en cerdos hospederos, el reordenamiento genético de los virus de influenza que infectan a aves, humanos y cerdos. Después de la transmisión al cerdo, los virus aviares y humanos sufren una evolución divergente y establecen las nuevas líneas genéticas (Vincent et al., 2009; Karasin et al., 2002; Webby et al., 2004).
El virus H3N2 doble recombinante se aisló por primera vez de los cerdos durante una severa enfermedad tipo influenza, que se presentó en las granjas de Carolina del Norte en 1988. Los análisis genéticos de ese virus revelaron que contenía segmentos provenientes del linaje porcino clásico (NS, NP, M, PB2 y PA), junto con algunos segmentos provenientes de las cepas humanas propagadas (HA, NA y PB1). Posteriormente, aislaron un virus H3N2 triple recombinante en cerdos en Minnesota, Iowa y Texas, que contenía segmentos genéticos procedentes del virus porcino clásico (NS, NP y M), del virus humano (HA, NA y PB1) y del virus aviar (PB2 y PA) (Karasin et al., 2006). Una característica constante en esta nueva reasociación entre diferentes tipos virales es la presencia de los genes internos específicos PB1, PB2, PA, NP, M y NS, a los que en conjunto se conoce como Genes Internos Triple Recombinantes (TRIG, por sus siglas en inglés), que en su origen derivaron de un virus H3N2 recombinante.
Desde 2005, se han identificado en cerdos canadienses y diseminado a través de Estados Unidos virus H1 tipo humano, que pertenecen a linajes genética y antigénicamente diferentes, derivados de un virus de influenza H1 clásico (Zhou et al., 1999; Gramer, 2008).
Los 6 genes internos en el virus H1 humano muestran semejanzas con los encontrados en los TRIG presentes en los recientes aislamientos virales. Esto sugiere que los virus que contienen los TRIG son capaces de aceptar múltiples tipos de genes HA y NA, lo que a su vez proporcionaría una ventaja de selección al virus porcino que los contuviera.
México ha publicado sólo un reporte acerca de un aislamiento viral porcino estrechamente relacionado con el virus A/H1N1 pdm09 (Escalera et al., 2012). Y no ha reportado ningún estudio sobre los cambios genéticos y la divergencia evolutiva de los genes internos PB2, PB1, PA, NP, M y NS1 del virus de influenza porcina que proliferó en la pandemia de 2009 y 2010. Así, el objetivo de este estudio fue identificar los cambios en estos genes y delinear su historia evolutiva.
Materiales y métodos
Recolección de muestras, aislamiento viral y extracción de ARN
Muestras de tejido pulmonar de cerdos recientemente sacrificados se recolectaron en 2009 y 2010 en rastros de 18 estados de México para un estudio previo y las muestras se mantuvieron en congelación. El Departamento de Medicina y Zootecnia de Cerdos (DMZC) de la Facultad de Medicina Veterinaria y Zootecnia (FMVZ) de la Universidad Nacional Autónoma de México (UNAM) realizó los ensayos de RT-PCR y el aislamiento viral en su laboratorio de bioseguridad nivel 3 (BSL3).
Cerdos de 7 de las 123 granjas inspeccionadas (5.6%) dieron positivo para el gen viral M. El RT-PCR se realizó utilizando el KIT OneStep RT-PCR QIAGEN®; se siguieron las instrucciones que indica el fabricante y se utilizaron los primers SIVM-F (5'TGAGTCTTCTAACCGAGGT 3') y SIVM-R (5' AGCGTCTACGCTGCAGTC R). Las condiciones de los ciclos para la amplificación de los genes fueron las siguientes: un ciclo a 50°C, durante 30 minutos; un ciclo de 95°C, durante 15 minutos; 35 ciclos a 94°C por 30 segundos, a 60°C por 60 segundos y a(¿) 72°C por 60 segundos, y un ciclo final de 72°C durante 10 minutos. Las muestras se dejaron a 4° por tiempo indefinido. Una vez que se obtuvo el producto de PCR, se mezclaron 5 µl del producto de PCR con 2 µl de buffer de muestra y se disolvió en gel de agarosa al 2% a 90 volts por 50 minutos en una cámara de electroforesis horizontal. El gel se tiñó con bromuro de etídio, y los amplicones esperados se visualizaron con un transiluminador Bio Imaging System y se identificaron mediante el uso de un "sistema de imagen Gel Logic 112" Kodak imager y el marcador de peso molecular pUC Mix 8®.
Para la replicación viral, se inocularon, en condiciones estériles, alícuotas de 200 µl en la cavidad alantoidea de embriones de pollo de 9 a 11 días de edad (ALPES1®) libres de patógenos específicos (SPF) e incubados a 37°C. El fluido alantoideo se recolectó a las 24, 48 y 72 horas posteriores a la inoculación y se centrifugó a 3500 rpm durante 5 minutos. Los títulos virales se evaluaron mediante la prueba de hemaglutinación. El ARN viral se extrajo utilizando el Kit PureLinkTM Viral RNA/DNA (Invitrogen, Carlsbad, CA) y se siguieron las instrucciones recomendadas por el fabricante. Después de extraer el ARN de los 7 virus aislados, la secuencia genómica completa de todos los aislamientos se determinó con el uso de una plataforma de secuenciación Illumina.
Secuenciación
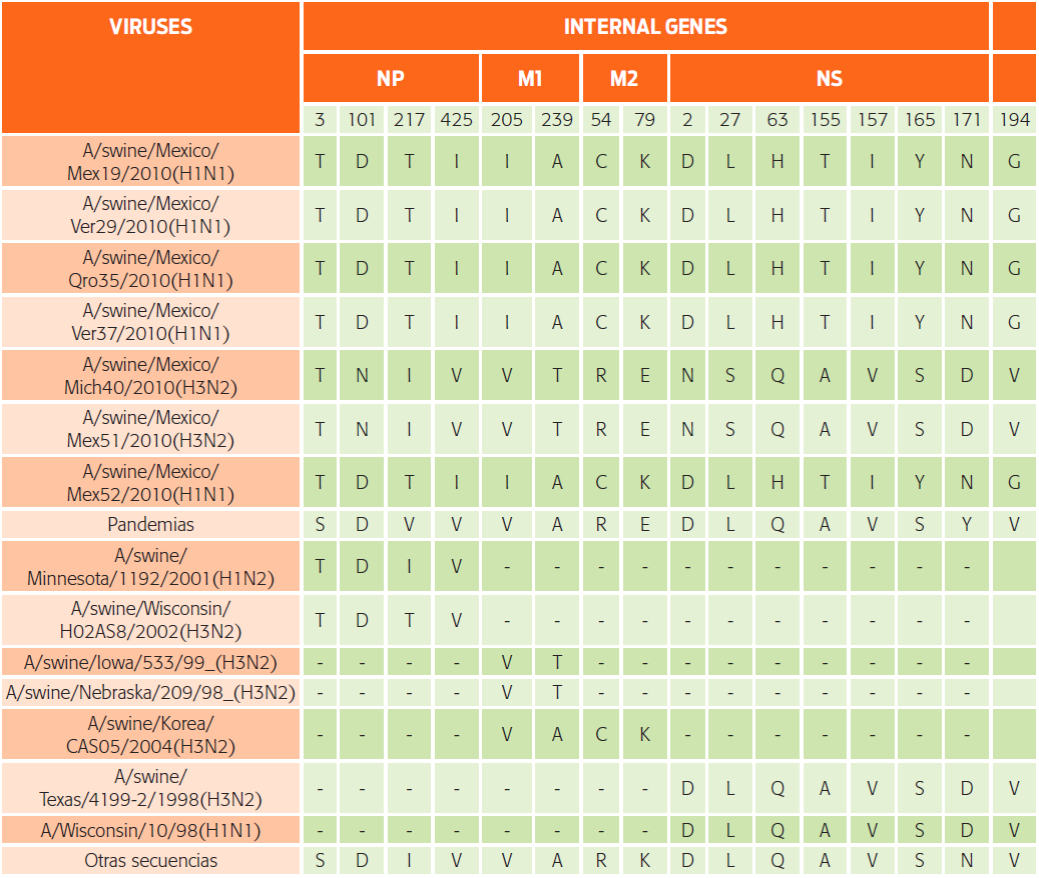
La secuenciación del genoma completo se realizó en el Instituto de Biotecnología (UNAM). Las librerías de 200 pares de bases se prepararon de acuerdo con el uso del Kit Illumina mRNA-Seq 8, del que se siguieron las instrucciones recomendadas por el fabricante, y se cargaron individualmente en el Genome Analyzer IIx (Illumina, San Diego, CA). La secuenciación se desarrolló en 36 ciclos de extensión de pares de bases simples. Para el análisis de imagen, se utilizó el Genome Analyzer Pipeline versión 1.4. Las lecturas virales se ensamblaron con MAQ version 0.7.1 (Mapping and Assembly with Qualities) (Li et al., 2008), utilizando el aislamiento de influenza A A/Netherlands/602/2009 como el genoma de referencia para el ensamble. Los gráficos de cobertura que coincidían con el genoma de referencia, generados mediante el uso de R2.12 (Team, 2010), sólo se considerarán para un análisis posterior. De cada muestra se obtuvieron entre 7.6 y 10.5 millones de lecturas de 36 nucleótidos de longitud. Más del 99.3% del genoma viral se secuenció con una cobertura de 120x a 1006x. Todas las secuencias se registraron en el GenBank y se encuentran disponibles en el National Center for Biotechnology (NCBI) (Cuadro 1).
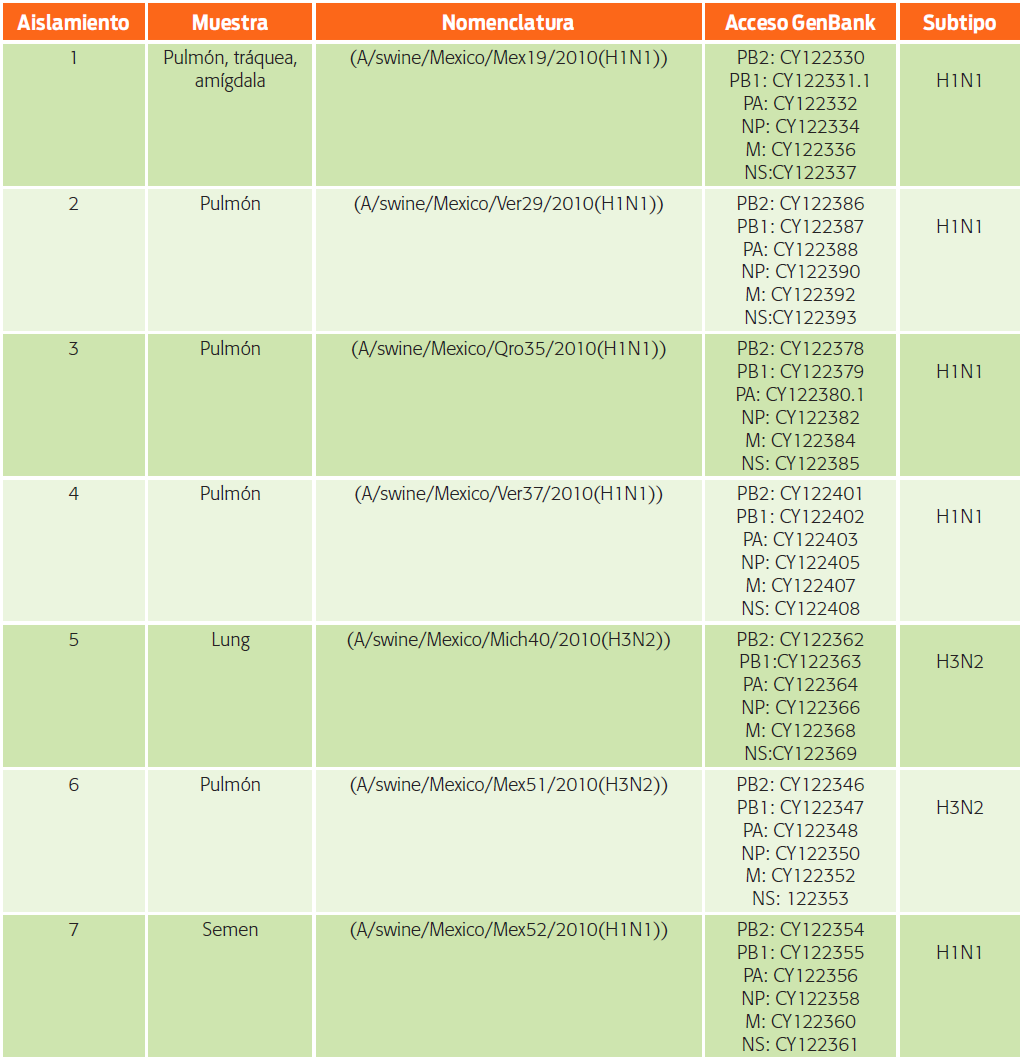
Análisis Filogenético
Una vez obtenidas las secuencias consenso, se realizó una búsqueda de identidad para una cobertura mayor al 95%, en la que se usó NCBI BLASTn (Basic Local Alignment Search Tool). Las secuencias resultantes se compararon con otras secuencias en el GenBank para la reconstrucción filogenética.
Las secuencias de referencia para el virus pandémico fueron A/Mexico/InDRE4487/2009 (H1N1), A/Mexico/LaGloria-3/2009 (H1N1), y A/MexicoCity/005/2009 (H1N1). Las secuencias utilizadas se muestran en el Cuadro 2.

La secuencia de la cepa viral A/Mexico/InDRE4487/2009 (H1N1) se elegió como referencia, debido a que fue reportada por el Instituto de Diagnóstico y Referencia Epidemiológica (InDRE), organismo acreditado para la vigilancia epidemiológica de la Red Nacional de Laboratorios de Salud Pública (RNLSP) en México. La cepa viral A/Mexico/LaGloria-3/2009 (H1N1) se seleccionó como uno de los aislamientos procedentes de la "zona cero" que dio origen a la pandemia del 2009. Finalmente, se incluyó el virus A/MexicoCity/005/2009 (H1N1) debido al reporte hecho por el Proyecto de Secuenciación del Genoma de Influenza (IGPS). La inclusión de las secuencias virales registradas en México puede ayudar a determinar si estos aislamientos porcinos comparten un origen ancestral común con los virus pandémicos en México. Las secuencias para cada gen se alinearon progresivamente en línea con el uso de MAFFT V.7 (programa de alineamiento múltiple para secuencias de aminoácidos o nucleótidos) (Kazutaka et al., 2002).
Para dilucidar las relaciones filogenéticas entre los genomas virales secuenciados, se utilizó el análisis JModelTest (Posadas D., 2008) junto con el criterio de máxima verosimilitud (ML).
El algoritmo se aplicó a cada conjunto de secuencias para determinar el mejor modelo de sustitución, junto con una prueba filogenética bootstrap de 1000 repeticiones para proporcionar apoyo estadístico en cada rama generada. Sólo los valores de bootstrap mayores o iguales a 0.80 (80%) se consideraron relevantes. Los árboles ML inferidos para la secuencia de nucleótidos de cada gen se construyeron utilizando diferentes modelos de sustitución, principalmente bajo el Criterio de Información Bayesiano (BIC):
Gen PB1 |
Hasegawa-Kishino-Yano (HKY) + Gamma Distributed (G) |
Gen PB2 |
Hasegawa-Kishino-Yano (HKY) + Gamma Distributed (G) |
Gen PA |
General Time Reversible (GTR) + Gamma Distributed (G) |
Gen NP |
Hasegawa-Kishino-Yano (HKY) + Gamma Distributed (G) |
Gen M |
Kimura 2-parameter (K2) + Gamma Distributed (G) |
Gen NS |
Hasegawa-Kishino-Yano (HKY) + Gamma Distributed (G) |
Para el análisis bioinformático de las secuencias PB1 y NS se utilizó el software MEGA (Molecular Evolutionary Genetics Analysis), versión 5.2 (Tamura et al., 2011). Los árboles filogenéticos se construyeron a partir de secuencias de nucleótidos y se aplicó el criterio bayesiano. A partir del modelo GTR, se generó un árbol filogenético para ambas secuencias.
Resultados y discusión
Caracterización filogenética de los genes internos PB2, PB1, PA, NP, M y NS
Con respecto a la estructura evolutiva de las topologías construidas para los genes PB2, NP y M, los virus pandémicos se unieron para formar grupos que resultaron totalmente diferentes a los cambios transitorios comunes de todos los virus porcinos incluidos en el análisis, el cual mostró que el gen PA de los virus pandémicos estudiados fue heredado de un ancestro común de origen aviar y divergió en los cerdos, lo que mantiene una línea de evolución continua (Fig. 1).
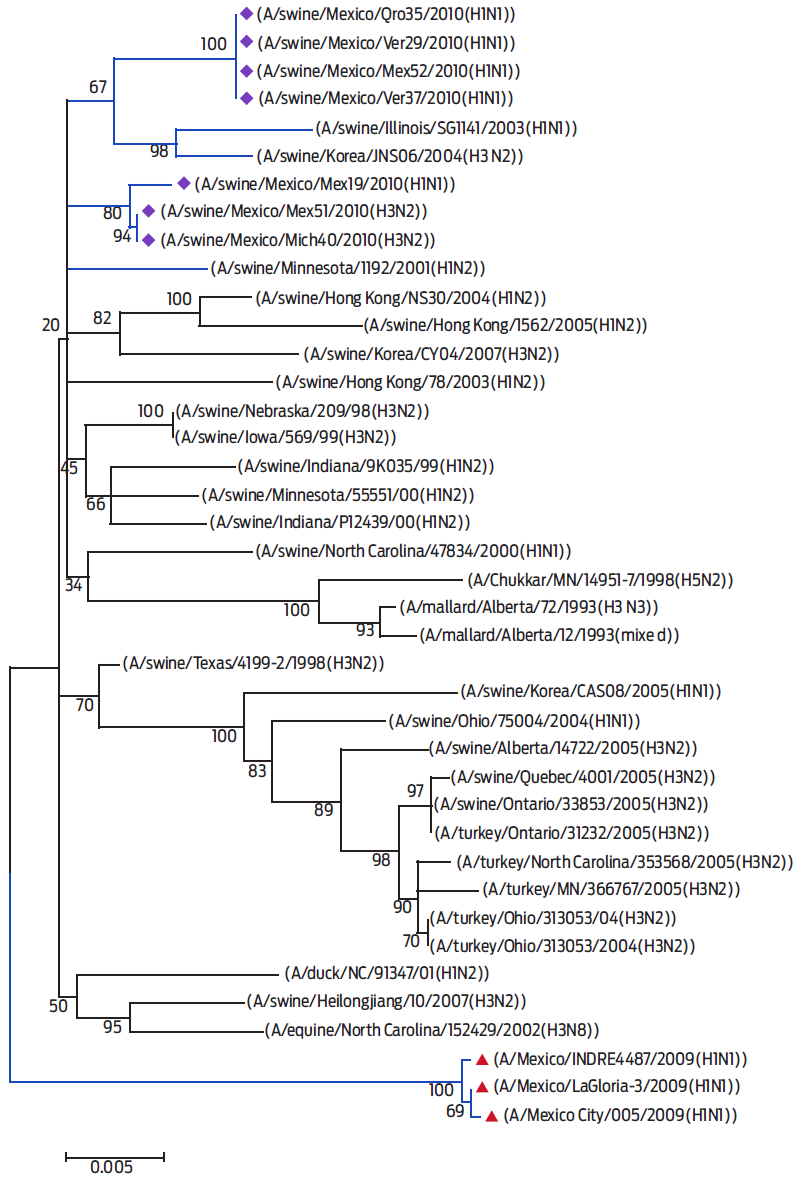
Figura 1 Análisis filogenético molecular por el método de Máxima Probabilidad (ML), utilizando el modelo de sustitución HKY+G. Árbol filogenético del gen PB2 de los 7 aislamientos virales (diamante violeta) y del virus pandémico (triángulo rojo) basado en la secuencia de nucleótidos y otras secuencias del GenBank. El árbol se dibujó a escala utilizando las mismas unidades de longitud de la rama y de la distancia evolutiva que se usaron para inferir el árbol filogenético. Las unidades de distancia evolutiva están en razón del número de sustituciones de bases por sitio.
Gen PB2
El análisis del gen PB2 (segmento 1) demostró que todos los virus aislados se distribuyeron a lo largo de una rama específica de genes de origen porcino después de la divergencia evolutiva entre las aves y los cerdos y están filogenéticamente relacionados con el virus A/swine/Minnesota/1192/2001 (H1N2). Los asilamientos 29, 35, 35 y 52 (H1N1), así como el 19 (H1N1) y el 52 (H3N2), divergieron del mismo ancestro pero bajo diferentes líneas evolutivas. Debido a que son diferentes subtipos virales, los aislamientos 19, 40 y 51 comparten un rasgo sinapomórfico (Fig.1).
En el gen PB2 se identificaron 6 cambios totales de aminoácidos exclusivos de los virus pandémicos respecto de todas las secuencias analizadas. Los aislamientos 29, 35, 37 y 52 mostraron cuatro cambios de aminoácidos en las posiciones I292M, V386A, V731I y L434F. Entre todas las secuencias alineadas, 2 de estos cambios se encontraron exclusivamente en estos virus, incluyendo los virus pandémicos. La posición 292 de la proteína PB2 se alineó con los virus A/swine/Illinois/SG1141/2003 (H1N1) y A/swine/Korea/JNS06/2004 (H3N2). Sin embargo, los aislamientos 19, 40 y 51 mostraron 4 cambios de aminoácidos individuales -no presentes en ninguna otra secuencia- en las posiciones A395V, I451T, L475M y T637A (Cuadro 3).

Gen PB1
Filogenéticamente, los aislamientos 19, 29, 35, 27 y 52 (H1N1) se incluyeron en un grupo monofilético con un rasgo sinafomórfico originado del virus americano A/swine/Nebraska/00188/2003 (H3N2) de diferentes subtipos, mientras los aislamientos 40 y 51 (H3N2) se agruparon por separado (Fig. 2)

Figura 2 Análisis filogenético molecular por el método de Máxima Probabilidad (ML), utilizando el modelo de sustitución HKY+G. Árbol filogenético del gen PB1 de los 7 aislamientos virales (diamante violeta) y del virus pandémico (triángulo rojo) basado en la secuencia de nucleótidos y otras secuencias del GenBank. El árbol se dibujó a escala utilizando las mismas unidades de longitud de la rama y de la distancia evolutiva que se usaron para inferir el árbol filogenético. Las unidades de distancia evolutiva están en razón del número de sustituciones de bases por sitio.
Se observaron 11 cambios de aminoácidos con respecto a las otras secuencias en el gen PB1 aislado del virus pandémico. Se encontró que los aislamientos 19, 29, 35, 37 y 52 (H1N1) tenían las mismas mutaciones en los aminoácidos K214R, I368V, D464N, T469A y F574Y, y que también tenían en común el residuo M744I con el virus A/swine/Nebraska/00188/2003 (H3N2). Los aislamientos 40 y 51 (H3N2) fueron las únicas secuencias que presentaron la mutación D565N (Cuadro 3).
Gen PA
La relación filogenética del gen PA (segmento 3) del virus A/swine/Mexico/Qro35/2010 (H1N1) con el virus de influenza humano estacional permitió identificar ramas evolutivamente heterogéneas dentro de un único grupo monofilético y una estrecha relación con el virus humano subtipo H3N2, cuyo valor bootstrap fue superior a100%. Se encontró que las secuencias de los genes PA de los otros aislamientos analizados están evolutiva y repetidamente relacionadas con el virus A/swine/Minnesota/1192/2001 (H1N2). Los aislamientos 40 y 51 (H3N2) mostraron homología sinapomórfica y se unieron en un grupo de genes de virus de cerdos y aves de corral con un ancestro común. Las secuencias en estos grupos muestran que divergen de los aislamientos 19, 29, 37 y 52 (H1N1). Estos grupos tienen un origen diferente al del virus de la estirpe americana (Fig. 3).
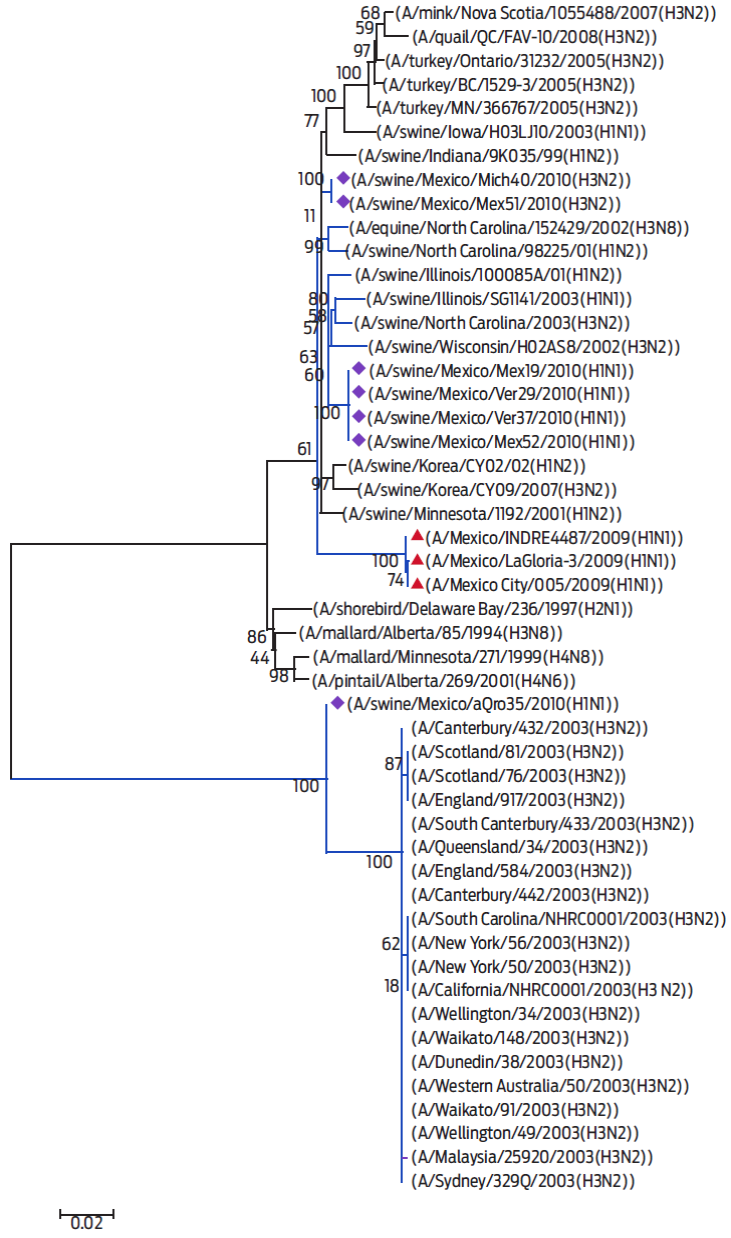
Figura 3 Análisis filogenético molecular por el método de Máxima Probabilidad (ML), utilizando el modelo de sustitución HKY+G. Árbol filogenético del gen PA de los 7 aislamientos virales (diamante violeta) y el virus pandémico (triángulo rojo) basado en la secuencia de nucleótidos y otras secuencias del GenBank. El árbol se dibujó a escala utilizando las mismas unidades de longitud de la rama y de la distancia evolutiva que se usaron para inferir el árbol filogenético. Las unidades de distancia evolutiva están en razón del número de sustituciones de bases por sitio.
Se detectaron 6 cambios de aminoácidos en esta proteína, pero sólo en la secuencia pandémica. Se identificaron 33 cambios de aminoácidos en el virus A/swine/Mexico/Qro35/2010 (H1N1) y todos ellos corresponden a secuencias de virus humanos. Por otra parte, se identificaron 3 cambios de aminoácidos en el aislamiento viral 35 (N254, R262 y R256) correspondientes a las secuencias virales pandémicas y humanas. Además, el asilamiento 35 mostró 2 mutaciones de aminoácidos (S259 y V459) que sólo se han encontrado en el virus A/Malaysia/25920/2003 (H3N2).
La variante en el aminoácido L403I sólo se encontró en los aislamientos 40 y 51, mientras que la variante R551K se observó en los aislamientos 19, 29, 35, 37 y 52. Los aislamientos virales 19, 29, 37 y 52 tienen un cambio de aminoácido en la posición Q344, diferente del aminoácido en esta posición en los aislamientos 35, 40 y 51 (L344). Se encontró que el cambio de aminoácidos en la posición V592 de los aislamientos 19, 29, 35, 37 y 52 era idéntico al de los virus A/swine/Illinois/SG1141/2003 (H1N1) y A/swine/Wisconsin/H02AS8/2002 (H3N2) (Cuadro 3).
Gen NP
Debido a que el gen NP juega un papel importante en la determinación de especificidad de la especie viral (Scholtissek et al., 1985), se hizo una caracterización completa de la filogenia de este gen (segmento 5). Los altos niveles de homología en los análisis BLASTn (98-100%) y BLASTp (98-100%) sugieren una muy cercana relación con los virus porcinos. Los 7 aislamientos analizados se aglutinaron en un grupo monofilético. Los asilamientos 40 y 51 (H3N2) se ubicaron en una rama evolutiva diferente como genes ortólogos con proximidad ancestral al virus A/swine/Minnesota/1192/2001 (H1N2), mientras que los asilamientos 19, 29, 35, 37 y 52 (H1N1) se reunieron en una rama aparte con proximidad ancestral al virus A/swine/Wisconsin/H02AS8/2002 (H3N2) (Fig. 4).
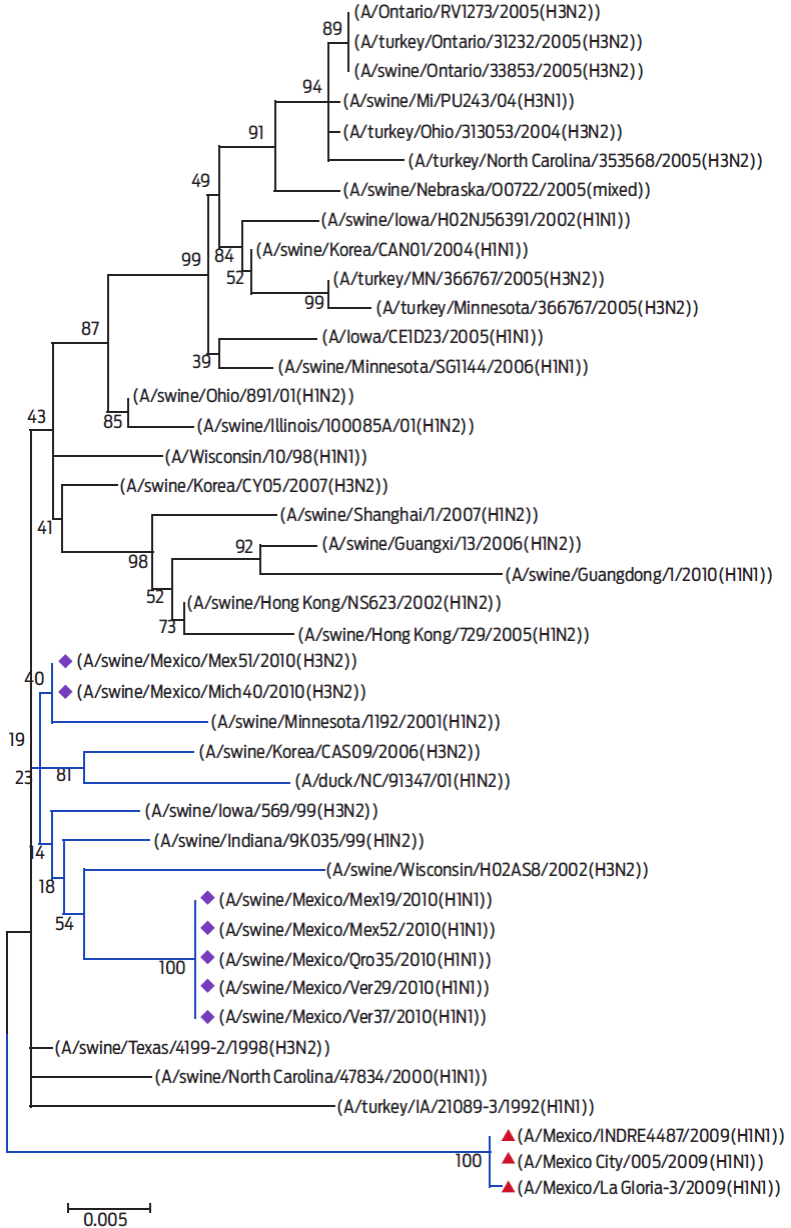
Figura 4 Análisis filogenético molecular por el método de Máxima Probabilidad (ML), utilizando el modelo de sustitución HKY+G. Árbol filogenético del gen NP de los 7 aislamientos virales (diamante violeta) y el virus pandémico (triangulo rojo) basado en la secuencia de nucleótidos y otras secuencias del GenBank. El árbol se dibujó a escala utilizando las mismas unidades de longitud de la rama y de la distancia evolutiva que se usaron para inferir el árbol filogenético. Las unidades de distancia evolutiva están en razón del número de sustituciones de bases por sitio.
Entre los aislamientos virales analizados, se identificaron 3 cambios insólitos de aminoácidos en el segmento NP de los aislamientos pandémicos y se encontró que la posición T3 de los aislamientos virales estudiados era la misma que en los aislamientos A/swine/Minnesota/1192/2001 (H1N2) y A/swine/Wisconsin/H02AS8/2002 (H3N2). Los aislamientos virales 40 y 51 mostraron un cambio distintivo en la posición V425I, como lo hicieron los aislamientos 19, 29, 35, 37 y 52 en la posición D101N. La mayoría de los cambios se observaron en la posición 217 (e.g. I217 y T217), aunque las secuencias pandémicas mostraron una variante V217 (Cuadro 4).
Gen M
Filogenéticamente, las secuencias del gen M son parálogas que divergieron de un ancestro que no suele basarse en el valor de la filogenia. Los aislamientos 40 y 51 (H3N2) están relacionados con virus porcinos y son cercanos al virus A/swine/Nebraska/209/98 (H3N2), con un valor de apoyo de bootstrap de 98%; en cambio, los aislamientos 19, 29, 37 y 52 (H1N1) se encuentran relacionados con el virus A/swine/Korea/CAS05/2004 (H3N2), con un valor de apoyo de bootstrap de 97% (Fig. 5). Se observaron 21 cambios de aminoácidos en el gen M de los virus pandémicos con respecto a las otras secuencias. El aminoácido en la posición A239T en la subunidad M1 se igualó únicamente con los aislamientos 40 y 51; mientras que V205I se igualó con los aislamientos 19, 29, 35, 37 y 52. La posición C4 en la subunidad M2 fue idéntica a la de los aislamientos 19, 29, 35, 37, 52 y A/swine/Korea/CAS05/2004 (H3N2). El aminoácido E479 sólo se igualó con las secuencias de los aislamientos 40, 51 y las secuencias pandémicas, mientras que K79 se igualó con los aislamientos 19, 29, 35, 37 y 52 (Cuadro 4).
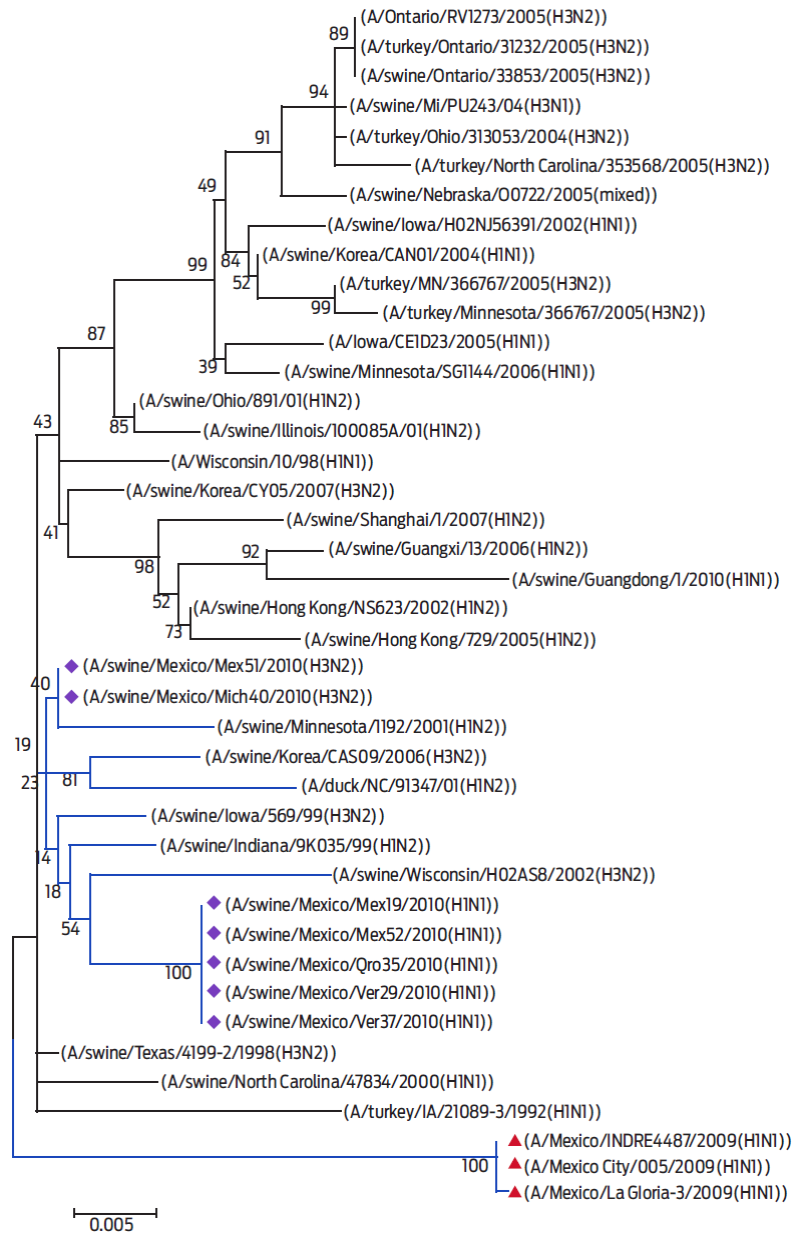
Figura 5 Análisis filogenético molecular por el método de Máxima Probabilidad (ML), utilizando el modelo de sustitución K2 + G. Árbol filogenético del gen M de los 7 aislamientos virales (diamante violeta) y el virus pandémico (triangulo rojo) basado en la secuencia de nucleótidos y otras secuencias del GenBank. El árbol se dibujó a escala utilizando las mismas unidades de longitud de la rama y de la distancia evolutiva que se usaron para inferir el árbol filogenético. Las unidades de distancia evolutiva están en razón del número de sustituciones de bases por sitio.
Gen NS
Una comparación de las secuencias de los genes NS sugiere que los aislamientos 19, 29, 35, 37 y 52 (H1N1) pertenecen a un grupo monofilético y que tienen una muy cercana relación sinamórfica con los virus americanos A/swine/NewMexico/SG1158/2003 (H3N2), A/swine/Oklahoma/00142/2003 (H3N2), y A/swine/Oklahoma/00142/2003 (H3N2), aunque éstos sean de diferentes subtipos. Por otra parte, los aislamientos 40 y 51 (H3N2) se ubicaron en grupos aparte, debido a que muestran una divergencia evolutiva del virus porcino A/swine/Texas/4199-2/1998 (H3N2) al virus humano A/Wisconsin/10/98 (H1N1), lo que da lugar a los aislamientos H3N2 mexicanos (Fig. 6).
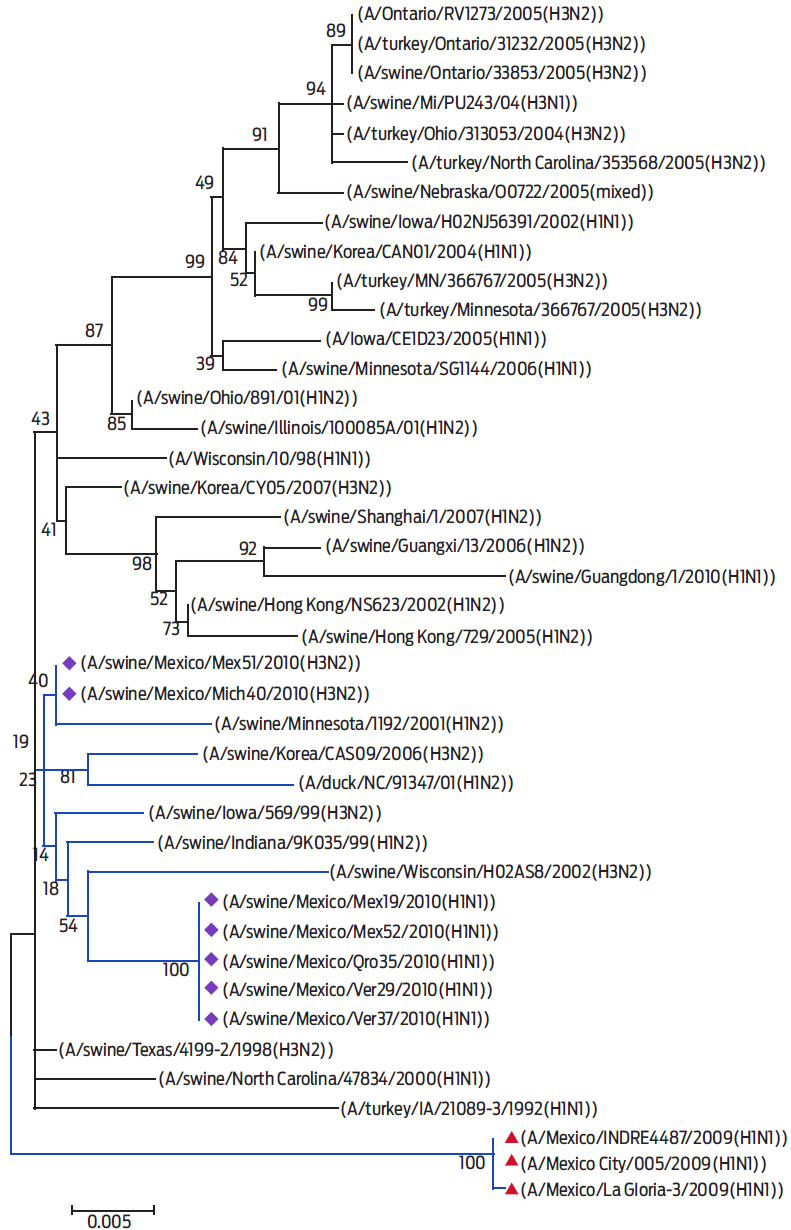
Figura 6 Análisis filogenético molecular por el método de Máxima Probabilidad (ML), utilizando el modelo de sustitución HKY + G. Árbol filogenético del gen NS de los 7 aislamientos virales (diamante violeta) y el virus pandémico (triangulo rojo) basado en la secuencia de nucleótidos y otras secuencias del GenBank. El árbol se dibujó a escala utilizando las mismas unidades de longitud de la rama y de la distancia evolutiva que se usaron para inferir el árbol filogenético. Las unidades de distancia evolutiva están en razón del número de sustituciones de bases por sitio.
Se observó un total de 9 cambios de aminoácidos en el gen NS de virus pandémicos, comparados con las otras secuencias.
Las variantes de los aminoácidos Q63H, A155T, V157I, S165Y y V194G se identificaron en los aislamientos 19, 29, 35, 37 y 52 (H1N1). L27S y D2N sólo se encontraron en los aislamientos 40 y 51 (H3N2), donde ambos comparten un cambio de aminoácido en la posición N171D con los asilamientos A/swine/Texas/4199-2/1998 (H3N2) y A/Wisconsin/10/98 (H1N1) (Cuadro 4).
La divergencia evolutiva activó la aparición de un número de cepas virales de influenza y permitió a los virus de la progenie establecerse y mantenerse en diferentes huéspedes. Distintos estudios han demostrado que el gen de la hemaglutinina es el principal gen viral involucrado en la elección del huésped. Sin embargo, los genes internos también juegan un papel importante en la exclusión del huésped (Webster et al., 1992).
La falta de esfuerzos sistemáticos de vigilancia y una estrategia de muestreo apropiado para el virus de la influenza en las poblaciones de cerdos en México, dificultaron la identificación oportuna de la pandemia de H1N1 en 2009, y la aparición de este virus que transporta los genes de los subtipos de Eurasia y América del Norte no fue identificado de inmediato.
En el presente estudio, la caracterización genética de los virus de influenza porcina aislados en México durante el 2009 y 2010, poco después de esta pandemia, mostró una relación filogenética completamente divergente con cepas de virus pandémicos. Sin embargo, el gen PA del virus A/swine/Mexico/Qro35/2010 (H1N1) mostró relación genética con el gen PA del virus estacional H3N2 de influenza humana. Esto sugiere una recombinación de virus de las especies porcina y humana, cuyas implicaciones son aún desconocidas.
A la reorganización de los genes en el complejo de las polimerasas, que algunas veces incluye al gen NP, se le ha involucrado en la atenuación de la virulencia o la replicación de la capacidad proliferativa en huéspedes específicos (Rott et al., 1979). En esta investigación también demostramos la existencia de 3 cambios de aminoácidos en el gen NP del virus pandémico, que sugiere una única diferencia con las cepas de virus no pandémicos. Escalera et al. (2012) demostraron la relación filogenética de los genes NP de los aislamientos virales porcinos provenientes de granjas de Querétaro, México, con 2 virus pandémicos, A/Mexico/La-Gloria-4/2009 (H1N1) y A/Mexico/LaGloria-8/2009 (H1N1), aislados de la "zona cero", lugar donde se identificó por primera vez la enfermedad. Sin embargo, los aislamientos obtenidos aquí, no contienen una nucleoproteína similar a la encontrada en los virus pandémicos. Un aislamiento del virus porcino proveniente del mismo estado de la República Mexicana (A/swine/Mexico/Qro35/2010 (H1N1)), como el analizado en nuestro estudio, es diferente del aislamiento reportado por Escalera et. al. (2012), quienes identificaron un polimorfismo en el aminoácido D53E, lo que sugiere que la cepa es más cercana al virus pandémico. Sin embargo, en esta investigación, el aislamiento (A/swine/Mexico/Qro35/2010 (H1N1)) no presenta este cambio, lo cual sugiere que se dio la transmisión humano-cerdo. Ali et. al. (2012) identificaron 4 mutaciones, G34A, D53E, I109T y V313I, en el gen NP de una recombinación entre los virus pandémicos y los porcinos H1N2. Nuestros aislamientos porcinos tienen una variación en V313F con respecto a la secuencia del virus pandémico. Estos cambios tienden a desempeñar un papel en la redistribución viral y la capacidad de adaptabilidad al huésped, como lo reportan Chen y Shih (2009) y Pan et. al. (2009). Ambos grupos de investigadores identificaron que el aminoácido en la posición 313 es crítico para la adaptación al huésped: Y313 para humanos, F313 para aves de corral y cerdos, y V313 para los virus pandémicos. El gen M confiere especificidad de especie (Scholtissek, 2002). Se ha informado que es posible distinguir las variantes del gen M de virus humanos y aviares por distintas sustituciones de aminoácidos en las proteínas M1 y M2. La mutación S31N del virus A/H1N1 pdm09 no se identificó durante la alineación de la secuencia de la subunidad M2 en el gen M en ninguna de las 7 cepas de la especie porcina reportadas por la CDC en la pandemia del 2009 (CDC, 2009). Estos virus contenían S31, una mutación conocida por conferir resistencia a los inhibidores del canal de protones M2 (amantadina y rimantadina). Sólo 3 secuencias pandémicas de referencia mantuvieron la mutación N31 (aunque también se identificaron las mutaciones en las posiciones 26, 27, 30, 31 y 34). Sin embargo, Weingartl et. al. (2011) observaron la misma mutación durante la caracterización genética de virus porcinos aislados en granjas canadienses, lo que confirma la transmisión del virus pandémico a cerdos. La presencia de brotes de A/H1N1 pdm09 en el cerdo y la transmisión directa de los virus humanos a cerdos fue referida por Moreno (2010) y Pereda (2010). No se detectó la mutación EG21, propuesta como relacionada con la adaptación al huésped.
Se ha señalado que el gen PB2 encontrado en los virus actuales de influenza porcina tiene un origen aviar. Sin embargo, aunque las secuencias porcinas mexicanas no están filogenéticamente relacionadas con los virus aviares, transportan la variante del aminoácido E627. En apariencia, E627 no se limita al gen PB2 aviar y permanece en el linaje del virus porcino triple recombinante. No se detectó el cambio al aminoácido K627, conocido porque concede adaptación a células humanas. Esto también fue descrito por Shu (2012) y Ali (2012), quienes encontraron que la única mutación que otorga adaptación a células humanas es V89M. La secuencia dada a conocer en este estudio muestra la presencia de residuos de valina en la posición 39 (V39).
Estos resultados indican que la secuencia con la mayor distancia evolutiva en México es el aislamiento A/swine/Mexico/Qro35/2010 (H1N1), que posee 33 aminoácidos idénticos a los del virus de influenza humana.
El aminoácido en la posición 552 es el principal sitio de regulación de la proteína PA, capaz de adaptar su funcionamiento en otras células huésped. De acuerdo con Mehle et. al. (2011), la mutación T552S en PA regula específicamente la adaptación del virus de influenza a las células huésped y generaría afinidad por las células humanas. Los autores sugieren que la mutación y recombinación del gen PA de virus aviar potenciarían la capacidad de infección del virus y el tropismo de especies. Sin embargo, con excepción de la secuencia A/swine/Mexico/Qro35/2010 (H1N1), que transporta la variante humana S552, los aislamientos porcinos aquí obtenidos tienen secuencias similares a las de los virus pandémicos y han mantenido sus rasgos aviares con respecto a la posición T552.
Aunque los genes internos PB1 y NS muestran variabilidad genética entre los actuales aislamientos porcinos, aún se desconoce si una mutación específica o incluso una serie de mutaciones contribuyen a la adaptación al huésped. Así, el papel de esos cambios de aminoácidos en la adaptación al huésped requiere investigarse. Se detectaron otras mutaciones patogénicas en estos genes. NS1 es responsable de suprimir la inducción del interferón (INF) antiviral durante la replicación viral. No se identificó la mutación D92E en cepas letales H5N1, que se ha relacionado con el incremento de la virulencia (Seo et al., 2002). Sin embargo, se ha demostrado que en los aislamientos porcinos de este estudio, los residuos K317 e I198 en la proteína PB1 -asociada con la patogenia en ratones- ha mutado a M317 y K198, respectivamente (Katz et al., 2000).
Por lo tanto, será vital el continuo monitoreo de la propagación de nuevas cepas de influenza porcina en México, así como estimar el riesgo de transmisión a otras especies. En China, se identificó un nuevo virus de influenza aviar H7N9 recombinante en febrero de 2013 (Gao et al., 2013). Cabe señalar que el primer brote de influenza pandémica en el siglo XXI se produjo por un nuevo virus de influenza H1N1 de origen porcino, que apareció a principios de 2009. Este virus es sustancialmente menos virulento que el virus de influenza de 1918, pero tiene el potencial de adquirir cambios de aminoácidos en proteínas virales clave que pueden incrementar su patogenia. Sin embargo, es importante explorar los factores por los que el huésped se involucra con la resistencia y susceptibilidad a la infección por el virus de influenza (Watanabe y Kawaoka, 2011).
Conclusiones
Nuestros análisis identificaron cambios en los genes de los virus de influenza porcina aislados en México en 2010, lo que sugiere que estas cepas de virus mantuvieron la distribución de los TRIG en los genes PB2, NP y M: un resultado no verificado con anterioridad en México. Los descubrimientos más relevantes en este estudio son la presencia de un gen PA del aislamiento viral A/swine/Mexico/Qro35/2010 (H1N1), derivado del virus humano H3N2, y la posibilidad de detectar nuevas cepas virales que incrementan el riesgo de trasmisión a otras especies. Es importante evaluar si los cambios de aminoácidos observados, modifican la patogenia del virus y la virulencia en los cerdos. Se ha demostrado que la población porcina puede actuar como reservorio de genes virales ancestrales de origen humano.
Financiamiento
Este estudio fue financiado por el Instituto de Ciencia y Tecnología, Proyecto Número PICSA 275/11.
REFERENCIAS
1 ) Ali A.; Khatri M, Wang L, Saif YM, and Lee CW. (2012). Identification of swine H1N2/pandemic H1N1 reassortant influenza virus in pigs, United States. Vet Microbiol. 158, 60-8. [ Links ]
2 ) CDC. Update: drug susceptibility of swine-origin influenza A (H1N1) viruses, April 2009. Morbidity and Mortality Weekly. Report 58, 433-435. [ Links ]
3 ) Chen, GW. and Shih SR, (2009). Genomic signatures of influenza A pandemic (H1N1) 2009 virus. Emerg. Infect. Dis. 15, 1897-1903. [ Links ]
4 ) Dawood, F.S.; Jain, S., Finelli, L., Shaw, M.W., Lindstrom, S., Garten, R.J., Gubareva, L.V., Xu, X., Bridges, C.B. and Uyeki, T.M. (2009). Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N. Engl. J. Med. 360, 2605-2615. [ Links ]
5 ) Escalera, Z.M.; Cobian-Guemes, G, Soto del Rio, M, Isa, P, Sanchez, B.I, Parissi, C.A, Martinez, C.M, Romero, P, Velazquez, S.L, Huerta, L.B, Nelson, M, Montero H, Vinuesa, P, Lopez, S, and Arias, C. (2012). Characterization of an influenza A virus in Mexican swine that is related to the A/H1N1/2009 pandemic clade. Virology 433, 176-182. [ Links ]
6 ) Gao, R. et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N. Engl. J. Med. 368, 1888-1897 (2013). [ Links ]
7 ) Garten, R.J.; Davis, C.T., Russell, C.A., Shu B, Lindstrom S, Balish A, Sessions WM, Xu X, Skepner E, Deyde V, Okomo-Adhiambo M, Gubareva L, Barnes J, Smith CB, Emery SL, Hillman MJ, Rivailler P, Smagala J, de Graaf M, Burke DF, Fouchier RA, Pappas C, Alpuche-Aranda CM, López-Gatell H, Olivera H, López I, Myers CA, Faix D, Blair PJ, Yu C, Keene KM, Dotson PD Jr, Boxrud D, Sambol AR, Abid SH, St George K, Bannerman T, Moore AL, Stringer DJ, Blevins P, Demmler-Harrison GJ, Ginsberg M, Kriner P, Waterman S, Smole S, Guevara HF, Belongia EA, Clark PA, Beatrice ST, Donis R, Katz J, Finelli L, Bridges CB, Shaw M, Jernigan DB, Uyeki TM, Smith DJ, Klimov A.I. and Cox NJ. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. (2009). Science. 325(5937):197-201 [ Links ]
8 ) Gramer, M.R. (2008). An update on swine influenza ecology and diagnostics. Proceeding, 39th Annual Meeting of the American Association of Swine Veterinarians. March 8-11, 2008, San Diego, CA, USA. [ Links ]
9 ) Karasin, A.I.; Landgraf, J., Swenson, S., Erickson, G., Goyal, S., Woodruff, M.,Scherba, G., Anderson, G. and Olsen, C.W. (2002). Genetic characterization of H1N2 influenza A viruses isolated from pigs throughout the United States. J. Clin. Microbiol. 40, 1073-1079. [ Links ]
10 ) Karasin, A.I.; Carman, S and Olsen CW. (2006). Identification of human H1N2 and human-swine reassortant H1N2 and H1N1 influenza A viruses among pigs in Ontario, Canada (2003 to 2005). J Clin Microbiol, 44, 1123-1126. [ Links ]
11 ) Katz, J.M., Lu, X., et al., 2000. Molecular correlates of influenza A H5N1 virus pathogenesis in mice. Journal of Virology 74 (22), 10807-10810. [ Links ]
12 ) Kazutaka K, Kazuharu M, Kei-ichi K, Takashi M. (2002). "MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform". Nucleic Acids Research 30 (14): 3059-66. doi:10.1093/nar/gkf436. PMC 135756. PMID 12136088. [ Links ]
13 ) Li, H, Ruan, J, Durbin, R. (2008). Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18, 1851-1858. [ Links ]
14 ) Mehle A.; Dugan, V, Taubenberger, J, and Doudna, J. (2011). Reassortment and Mutation of the Avian Influenza Virus Polymerase PA Subunit Overcome Species Barriers Journal of Virology. 1750-1757. [ Links ]
15 ) Moreno, A; Trani, D.L, Alborali, L, Vaccari, G, Barbieri, I, Falcone, E, Sozzi, E, Puzelli, S, Ferri, G, and Cordioli P. (2010). First pandemic H1N1 outbreak from a pig farm in Italy. Open Virol. J. 4, 52-56 [ Links ]
16 ) Pan C.; Cheung B, Tan S, Li C, Li L, Liu S, and Jiang S, (2009). Genomic signature and mutation trend analysis of pandemic (H1N1) 2009 influenza A virus. PLoS One 5, e9549. [ Links ]
17 ) Pereda, A.; Rimondi, A, Cappuccio, J, Sanguinetti, R, Angel, M, Ye J, Sutton, T, Dibarbora M, Olivera, V, Craig, MI, Quiroga, M, Machuca, M, Ferrero, A, Perfumo, C, and Perez DR. (2011). Evidence of reassortment of pandemic H1N1 influenza virus in swine in Argentina: are we facing the expansion of potential epicenters of influenza emergence? Influenza Other Respir. Viruses 5, 409-412. [ Links ]
18 ) Posada D. (2008). JModeltest: Phylogenetic Model Averaging. Molecular Biology and Evolution 25: 1253-1256. [ Links ]
19 ) Rott R.; Orlich M, and Scholtissek C. (1979). Correlation of pathogenicity and gene constellation of influenza A viruses. Non-pathogenic recombinants derived from highly pathogenic parent strains. J. Gen. Virol. 44, 471-477. [ Links ]
20 ) Scholtissek C.; Stech J, Krauss S, and Webster RG, (2002). Cooperation between the hemagglutinin of avian viruses and the matrix protein of human influenza A viruses. J. Virol. 76, 1781-1786. [ Links ]
21 ) Scholtissek, C.; Buerger, H., Kistner, O, and Shortridge K. F. (1985). The nucleoprotein as a possible major factor in determining hosts specificity of influenza H3N2 viruses. Virology 147, 287-294. [ Links ]
22 ) Seo S, H, Hoffmann E, Webster R, G (2002) Lethal H5N1 influenza viruses escape host anti-viral cytokine responses. Nat Med 8: 950-954. [ Links ]
23 ) Shu, B.; Garten, R, Emery, S, Balish A, Cooper, L, Sessions, W, Deyde, V, Smith C, Berman, L, Klimov, A, Lindstrom, S, and Xu X. (2012). Genetic analysis and antigenic characterization of swine origin influenza viruses isolated from humans in the United States, 1990-2010. Virology 422. 151-160. [ Links ]
24 ) Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22, 2688-2690. [ Links ]
25 ) Tamura, K, Peterson, D, Peterson, N, Stecher, G, Nei M, and Kumar, S. (2011). MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance and Maximum Parsimony Methods. Mol. Biol. Evol. 28, 2731-2739. [ Links ]
26 ) Tang J.W, Shetty N, Lam T.T, Hon K.L. (2010). Emerging, novel, and known influenza virus infections in humans. Infect Dis Clin North Am. 24:603-617. [ Links ]
27 ) Team, R. (2010). A language and Environment for Statistical Computing, R Foundation for Statistical Computing Vienna. [ Links ]
28 ) Tong S, Zhu X, Li Y, Shi M, Zhang J, et al. (2013) New World Bats Harbor Diverse Influenza A Viruses. PLoS Pathog 9(10): e1003657. doi:10.1371/journal.ppat.1003657. [ Links ]
29 ) Vincent, A.L.; Ma, K. M. Lager, B.H. Janke, and J.A. Richt. (2008). Swine Influenza Viruses a North American perspective. Adv. Virus. Res. 72,127-54. [ Links ]
30 ) Vincent A.L,; Swenson, S.L., Lager K.M, Gauger, P.C, Loiacono, C, and Zhang Y. (2009). Characterization of an influenza A virus isolated from pigs during anoutbreak of respiratory disease in swine and people during acounty fair in the United States. Veterinary Microbiology 137, 51-59. [ Links ]
31 ) Watanabe T, Kawaoka Y. (2011) Pathogenesis of the 1918 Pandemic Influenza Virus. PLoS Pathog 7(1): e1001218. doi:10.1371/journal.ppat.1001218. [ Links ]
32 ) Webster R.; Bean W, Gorman O, Chambers T, and Kawaoka Y. (1992). Evolutionand Ecology of Influenza A Viruses. Microbiological Reviews. 56, 152-179. [ Links ]
33 ) Webby, R.J.; Rossow, K., Erickson, G., Sims, Y. and Webster, R., (2004). Multiplelineages of antigenically and genetically diverse influenza Avirus co-circulate in the United States swine population. Virus. Res. 103, 67-73. [ Links ]
34 ) Weingartl, H.M.; Berhane, Y, Hisanaga, T, Neufeld, J, Kehler, H, Emburry-Hyatt C, Hooper-McGreevy, K, Kasloff, S, Dalman, B, Bystrom, J, Alexandersen, S, Li Y, and Pasick J. (2010). Genetic and pathobiologic characterization of pandemic H1N1 2009 influenza viruses from a naturally infected swine herd. J Virol. 84: 2245-56. [ Links ]
35 ) Zhou, N.N.; Senne, D.A, and Landgraf, J.S. (1999). Genetic reassortment of avian, swine, and human influenza A viruses in American pigs. J Virol, 73, 8851-8856. [ Links ]
Recibido: 17 de Enero de 2014; Aprobado: 19 de Junio de 2014
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
