











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Due to their high biocompatibility, high bioactivity, customized sequences and functionalities, flexible self-assembly ability, and biomimetic features, peptides have been widely exploited in materials science, nanotechnology, analytical science, biomedicine, tissue engineering, and other domains. Because peptides have a high affinity for some nanomaterials (such as inorganic nanoparticles, nanotubes, graphene, and other two-dimensional materials), the peptide- functionalized materials are biocompatible and bioactive, allowing them to be used in a variety of biomedical applications such as biosensors, cancer therapy, tissue engineering, and targeted drug delivery (Ulijin and Jerala, 2018).
Peptides with specific motifs can also be employed to facilitate the biomimetic production of inorganic nanomaterials, which can be used to make functional nanodevices for biosensing, energy storage, and environmental science. Furthermore, the self-assembly capacity of peptide molecules can be induced to form diverse superstructures, such as peptide nanospheres, nanofibrils, nanosheets, and hydrogels, based on the tailoring of peptide sequences (Lampel et al., 2018).
Cyclic peptides have several desirable qualities, including high binding affinity, target selectivity, and low toxicity, which make them a promising therapeutic development approach. Antimicrobial peptides (AMPs), also known as host defense peptides, are short, positively charged peptides found in a wide range of life forms, including microbes and humans. The majority of AMPs are capable of directly killing microbial infections, whereas some operate indirectly by altering the host defensive mechanisms (Mahlapuu et al., 2016).
Despite the fact that many studies have been conducted in this promising research subject, more research into the nanochemistry and nanotechnology of peptides is still necessary and crucial. As a result, we believe it is critical to investigate the chemical reactivity, which may aid in the development of various medicines through the use of tools provided by molecular modeling and computational chemistry. At present, in molecular modeling and computational chemistry, we recognize conceptual density functional theory (DFT) (Chattaraj et al., 2009; Gázquez et al., 2007; Geerlings et al., 2003 and 2020; Parr and Yang, 1989). as the most powerful tool that is currently available for studying the chemical reactivity of molecular systems. Conceptual DFT, which is also known as chemical reactivity theory, is capable of predicting the way in which chemical reactions take place by applying a series of global and local descriptors (Ayers and Parr, 2000; Poater et al., 2009 and 2010).
Cyclosporin A is a cyclic peptide of eleven amino acid residues which work as an immunosuppressant very useful for its clinical property in organ transplantation. It was initially isolated as an antifungal agent from several fungi, including Tolypocladium inflated. Alisporivir, a non-immunosuppressive cyclosporin A-analogue, inhibited MERS-CoV replication in vitro and it is being considered as a therapeutic option for the treatment of the COVID-19 infection (Pawlotsky, 2020).
With the conviction that the knowledge of the chemical reactivity is essential for the development of new medicines, we are reporting the results of a computational nanochemistry study of the chemical reactivities and bioactivity properties of two antimicrobial peptides using a CDFT-based computational peptidology (CDFT-CP) methodology (Flores-Holguín et al., 2020a, b, c; 2021a, b), which is derived from the combination of the chemical reactivity descriptors derived from conceptual density functional theory (CDFT) and some cheminformatics tools useful in the design of therapeutic drugs (Bajorath, 2014; Begam and Kumar, 2012; Benjamin, 2015; Engel and Gasteiger, 2018a, b; Guha and Bender, 2012; Medina-Franco and Saldívar-González, 2020; Varnek and Tropsha, 2008). This is complemented by an examination of the bioactivity and pharmacokinetics indices of the peptides in relation to the ADMET (absorption, distribution, metabolism, excretion, and toxicity) features. These findings provide further evidence of the superiority of the MN12SX density functional in fulfilling the Janak and ionization energy theorems using an earlier proposed KID methodology for validation. This has proven to be beneficial in accurately predicting CDFT indices, which is of help in the understanding of the chemical reactivity. The computational pharmacokinetics study revealed the potential ability of both cyclopeptides as therapeutic drugs through the interaction with different target receptors. The ADMET indices confirmed this assertion through the absence of toxicity and good absorption and distribution properties (Chakraborty et al., 2012; Daina et al., 2017; Pires et al., 2015).
Graphical sketches of the molecular structures of alisporivir and cyclosporin A are displayed in Figure 1:
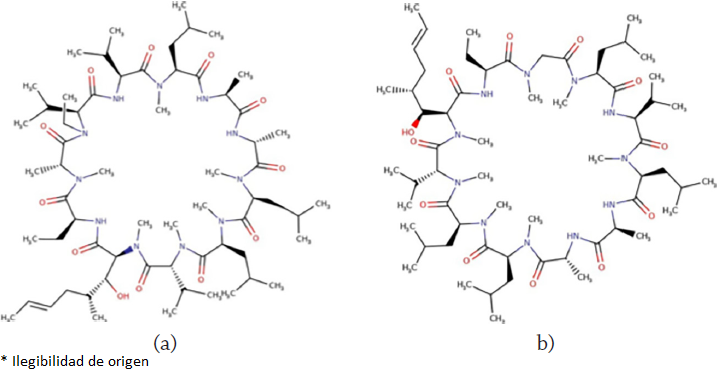
Theoretical background and computational details
Density functional theory (DFT) calculations
The Kohn-Sham (KS) methodology involves the electronic density, the determination of the molecular energy, and the orbital energies of a specific system, in particular, the HOMO and LUMO frontier orbitals which are intrinsically related to the chemical reactivity of the molecules (Cramer, 2004; Jensen, 2007; Lewars, 2003; Young, 2001). The definitions for the global reactivity descriptors are (Chattaraj et al.2009; Gázquez et al., 2007; Geerlings et al., 2003 and 2020; Parr and Yang, 1989):
being EH and EL the frontier orbital energies related to the cyclopeptides considered in this research. These global reactivity descriptors that arise from conceptual DFT (Chattaraj et al.; Gázquez et al., 2007; Geerlings et al., 2003 and 2020; Parr and Yang, 1989), have been complemented by the nucleophilicity index N (Domingo et al., 2008; Domingo and Pérez, 2011; Domingo et al., 2016; Domingo and Sáez, 2009; Jaramillo et al., 2008), that takes into account the value of the HOMO energy obtained by means of the KS scheme using an arbitrary shift of the origin with tetracyanoethylene (TCE) as a reference.
The conformers of both antimicrobial peptides were established using Marvin view 17.15 from ChemAxon [http://www.chemaxon.com], which was applied in order to undertake molecular mechanics calculations utilizing the MMFF94 force field with an energy window of 0.1 kcal/mol (Halgren, 1996 a,b,c and 1999; Halgren and Nachbar, 1996). This was followed by a geometry optimization and frequency calculation using the density functional tight binding (DFTB) methodology (Frisch et al., 2016). This last step was required for the verification of the absence of imaginary frequencies to confirm the stability of the optimized structure as being a minimum in the energy surface. The determination of the electronic properties and the reactivity descriptors of the cyclopetides are addressed through the MN12SX/Def2TZVP/ H2O model chemistry (Peverati and Truhlar, 2012; Weigend, 2006; Weigend and Ahlrichs, 2005), because it has been previously shown that it verifies the KID procedure and satisfies the ionization energy theorem (Flores-Holguín et al., 2020a, b, c; 2021a, b), with the aid of the Gaussian 16 software Frisch et al., 2016) and the SMD solvation model (Marenich et al., 2009). The charge of both molecules is taken equal to zero whereas the radical anion and cation are considered in the doublet spin state. The SMD solvation model was chosen because it has been shown that it provides atomic charges of the Hirshfeld type that are almost independent of the basis set, and which are usually recommended for calculations within conceptual density functional theory.
Computational pharmacokinetics and ADMET report
The SMILES notation of the cyclopeptides were acquired by accessing the PubChem database [https://pubchem.ncbi.nlm.nih.gov/], and then were fed into the online program chemicalize from ChemAxon [http://www.chemaxon.com], which was considered to get a glimpse of the potential therapeutic properties of the studied molecular systems (date of access: January 2022).
A similarity search in the chemical space of compounds with molecular structures that could be compared to the ones being studied, with already known biological and pharmacological properties, was achieved through the online Molinspiration software from Molinspiration Cheminformatics [https://www.molinspiration.com/] (accessed, January 2022). SwissTargetPrediction is an online tool for the prediction of protein targets of small compounds, and it was used to estimate the potential bioactivity of the antimicrobial pep- tides studied in this research (Daina et al., 2019).
Pharmacokinetics is a procedure that involves determining the likely fate of a medicinal molecule in the body, which is critical information in the creation of a new medicine. Individual indices named absorption, distribution, metabolism, excretion, and toxicity (ADMET) factors have typically been used to analyze the associated consequences. Chemicalize and the internet available SwissADME program were used to estimate some ADMET parameters in this study (Daina et al., 2017). pkCSM, a software for the prediction of small-molecule pharmacokinetic properties using SMILES that can be accessed through its linked webpage, was also used to obtain additional information regarding the Pharmacokinetics parameters and ADMET indices (Pires et al., 2015).
Results and discussion
The optimized molecular structures of the alisporivir and cyclosporin A antimicrobial peptides considered through this research using the methodology presented before are displayed in Figure 2:
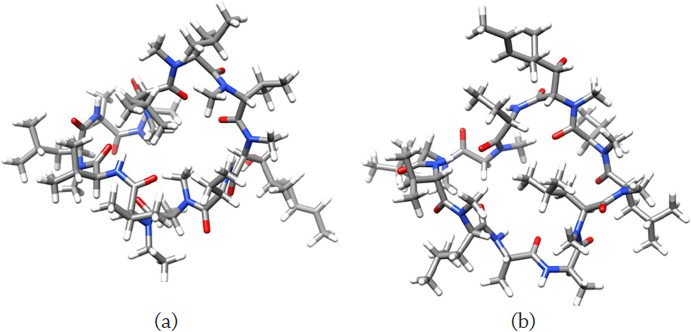
Source: Author’s elaboration.
Figure 2 Optimized molecular structures of: a) alisporivir and b) cyclosporin A.
The quality of the chosen density functional may be realized by comparing its results with results from high-level computations or from experimental values. Nevertheless, this comparison is not always computationally practicable because of the large size of the molecules or the lack of for the chemical methods being explored. Our research group has developed a methodology known as KID (Flores-Holguín et al., 2020a, b, c; 2021a, b), as an aid to evaluate a particular density functional with regard to its internal coherence. It is evident that within the generalized Kohn-Sham (GKS) version of DFT, some relationships exist between the KID methodology and the ionization energy theorem, which is a corollary of Janak theorem (Janak, 1978; Kanchanakungwankul and Truhlar, 2021; Kar et al., 2013; Tsuneda and Hirao, 2014; Tsuneda et al., 2010. This is done by connecting EH to -I and EL to -A, through
Another KID descriptor ∆SL related to the difference in energies between the SOMO and the LUMO of the neutral system has been devised to aid in the verification of the accuracy of the methodology.
The MN12SX density functional has been shown to have a Koopmans-compliant behavior in earlier studies of the chemical reactivity of diverse molecular systems (Flores-Holguín et al., 2020a, b, c; 2021a,b). However, for a further validation of this model chemistry in the prediction of the chemical reactivity properties of the antimicrobial peptides considered here, additional research is necessary. The CDFT software tool was used to make this determination, and the findings are shown in Table 1.
Table 1 HOMO, LUMO and SOMO orbital energies, HOMO-LUMO gap and the KID descriptors (all in eV) tested in the verification of the Koopmans-like behavior of the MN12SX density functional for the alisporivir and cyclosporin A cyclopeptides.
| Molecule | HOMO | LUMO | SOMO | H-L Gap | J(I) | J(A) | J(HL) | ∆SL |
|---|---|---|---|---|---|---|---|---|
| Alisporivir | -6.4594 | -1.0123 | -1.0065 | 5.4472 | 0.026 | 0.001 | 0.026 | 0.006 |
| Cyclosporin A | -6.5013 | 0.8199 | -0.7910 | 5.6815 | 0.011 | 0.012 | 0.016 | 0.029 |
Source: Author’s elaboration.
The results from Table 1 are very interesting because they show that there is an almost perfect fulfillment of the Janak and ionization energy theorems for the MN12SX/Def2TZVP/H2O model chemistry employed in this work.
Having verified that the MN12SX/Def2TZVP/H2O is the most adequate one for obtaining accurate results for the conceptual DFT global reactivity descriptors, the estimated values for the global reactivity descriptors (including the nucleophilicity N) for the three molecular systems acquired utilizing the mentioned CDFT tool are displayed in Table 2:
Table 2 Global reactivity descriptors for the alisporivir and cyclosporin A cyclopeptides: electronegativity (χ), hardness (η), electrophilicity (ω) (all in eV), softness S (in eV−1), nucleophilicity N, electrodonating power (ω−), elctroaccepting power (ω+) and net electrophilicity (∆ω±) (also in eV).
| Molecule | χ | η | ω | S | N | ω- | ω+ | ∆ω |
|---|---|---|---|---|---|---|---|---|
| Alisporivir | 3.7359 | 5.4472 | 1.2811 | 0.1836 | 2.3331 | 4.7705 | 1.0347 | 5.8052 |
| Cyclosporin A | 3.6606 | 5.6815 | 1.1793 | 0.1760 | 2.2912 | 4.5440 | 0.8833 | 5.4273 |
Source: Author’s elaboration.
The electronegativity (χ) and global hardness (η) are absolute values for the chemical reactivity that have not a known experimental counterpart. Indeed, they can be estimated by resorting to the experimental vertical ionization energy (I) and vertical electron affinity (A) but these values are not known for the molecule under study. A different thing can be said about the electrophilicity ω and the nucleophilicity (N). The electrophilicity ω index involves a compromise between the tendency of an electrophile to acquire extra electron density and its resistance to exchange electron density with the environment (Domingo et al., 2016). By considering a group of Diels-Alder reactions and the electrophiles involved in them (Domingo et al., 2002; Domingo and Sáez, 2009; Pérez et al., 2003), a classification of organic compounds as strong, moderate, or marginal electrophiles, that is an electrophilicity ω scale, was established, with ω larger than 1.5 eV for the first instance, with ω between 0.8 and 1.5 eV for the second case, and ω smaller than 0.8 eV for the final case (Domingo et al., 2002; Domingo and Sáez, 2009; Pérez et al., 2003). By checking Table 2, it can be said that both antimicrobial peptides may be regarded as moderate electrophiles. Domingo and his collaborators (Domingo et al., 2008; Domingo and Pérez, 2011; Domingo et al., 2016; Domingo and Sáez, 2009; Jaramillo et al., 2008), have also proposed a nucleophilicity index N through the consideration of the HOMO energy obtained through the KS scheme with an arbitrary shift of the origin taking the molecule of tetracyanoethylene (TCE) as a reference. An analysis of a series of common nucleophilic species participating in polar organic reactions allowed them to establish a further classification of organic molecules as strong nucleophiles with N > 3.0 eV, moderate nucleophiles with 2.0 <N < 3.0 eV and marginal nucleophiles with N < 2.0 eV. By checking again Table 2, it can be concluded that both molecules may be considered also as moderate nucleophiles.
The global descriptors demonstrate the chemical reactivity of each molecule in its entirety; therefore, local reactivity descriptors have been designed to assess the differences in the chemical reactivity between the areas inside a molecule. The nucleophilic and electrophilic Fukui functions (NFF and EFF) (Geerlings et al., 2003; Parr and Yang, 1989) and the dual descriptor DD (Martínez-Araya, 2012a, b, and 2015; Morell et al., 2005 and 2006; Toro-Labbé, 2007) are some of these local reactivity descriptors. They have defined as: NFF = ρN +1(r ) − ρN (r), EFF = ρN (r) − ρN−1(r) and DD = (∂ f (r )/∂ N)υ(r) , establishing links between the electronic densities of the various species as well as between the NFF and EFF. The NFF identifies molecular locations that are more vulnerable to nucleophilic attacks, whereas the EFF identifies regions that are more vulnerable to electrophilic attacks. The reactive locations have been successfully identified using these local reactivity characteristics. However, the DD has been discovered to be capable of describing both nucleophilic and electrophilic locations within a molecule without ambiguity (Martínez-Araya, 2015). Figure 3 shows graphical sketches of the DD for alisporivir and cyclosporin A, highlighting the locations where DD > 0 and DD < 0 for a better understanding of these molecules’ local chemical reactivity.
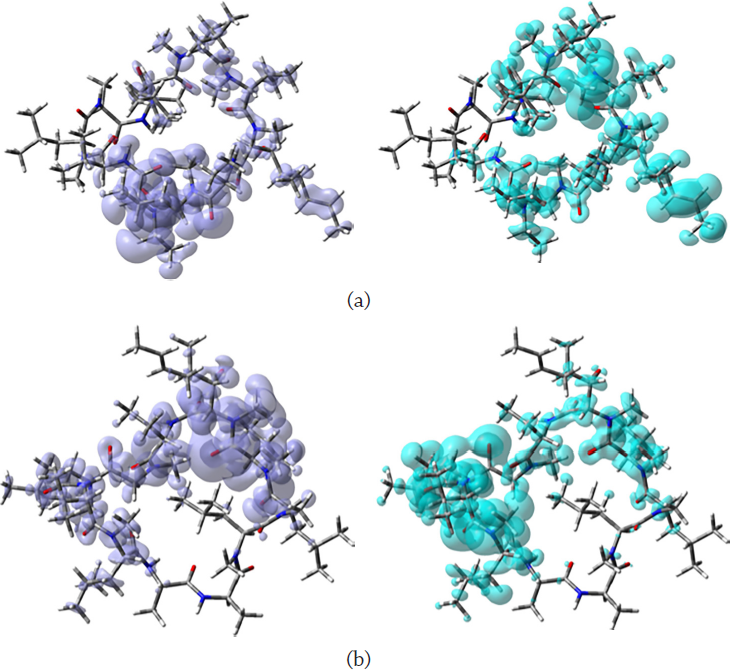
Source: Author’s elaboration
Figure 3 Graphical representation of the DD of the a) alisporivir and b) cyclosporin A molecules: left: DD > 0, right: DD < 0.
Computational pharmacokinetics and ADMET report
On the basis of the methodology presented previously, the pKas of both cyclopeptides have been estimated following a simple QSAR relationship pKa = 16.3088 - 0.8268 η that we have derived during the study of amino acids and small peptides, and which has been useful in the study of larger peptides as well as being of interest for the development of advanced glycation end products (AGEs) inhibitors (Frau et al., 2017). These values together with some results that can be potentially useful for future QSAR studies are reported in Table 3.
Table 3 Predicted parameters useful for QSAR studies for the alisporivir and cyclosporin A cyclopeptides: ∆G of solvation (in Kcal/mol), pKa, logP, TPSA (Å2) and molecular volume (Å3).
| Molecules | ∆G of solvation | pKa | logP | TPSA | Molecular volume |
|---|---|---|---|---|---|
| Alisporivir | -50.58 | 11.81 | 3.79 | 278.78 | 1219.70 |
| Cyclosporin A | -47.66 | 11.61 | 3.61 | 278.78 | 1203.12 |
Source: Author’s elaboration.
Although this research deals with the use and validation of certain computational techniques applied in the determination of the chemical reactivity properties of the studied molecules, it would be desirable to find some correlation between the conceptual DFT descriptors and the pharmacokinetics and ADMET indices as it has been shown for the case of the pKas. However, there is no sense in finding QSAR relationships when working with only two molecules. Some qualitative correlations can be mentioned instead. For example, logP for alisporivir is greater than for cyclosporin A and this correlates with the calculated values of the electrophilicity ω. The same conclusions can be obtained for the TPSA and the ∆G of solvation. Indeed, this approximate correlation is also found with the inverse of the chemical hardness η. This paves the way for considering these reactivity descriptors as an indication of their bioactivity when considering a larger number of molecular species.
The pharmacokinetics of a drug is evaluated through ADMET research, which is an acronymous for absorption, distribution, metabolism, excretion, and toxicity. If absorption is unsatisfactory, the distribution and metabolism of the drug would be changed, potentially resulting in nephrotoxicity and neurotoxicity. As a result, ADMET analysis is one of the most important aspects of computational drug design. In addition to the previous conceptual DFT-based computational peptidology results, we are supplementing this study with a report of the computed ADMET features as shown in Table 4.
Table 4 Absorption, distribution, metabolism, excretion and toxicity (ADMET) parameters related to alisporivir and cyclosporin A pharmacokinetics.
| Paramstsr | Alisporivir | Cyclosporin A |
|---|---|---|
| Absorption | ||
| Water solubility (log mol/L) | -2.892 | -2.892 |
| Caco2 permeability (log Papp 10-6 cm/s) | 1.699 | 1.716 |
| Intestinal absorption (human) (% absorbed) | 0 | 0 |
| Skin permeability (log Kp) | -2.735 | -2.735 |
| P-glycoprotein substrate | Yes | Yes |
| P-glycoprotein I inhibitor | Yes | Yes |
| P-glycoprotein II inhibitor | No | No |
| Distribution | ||
| VDss (human) ( log L/kg) | 0.027 | 0.030 |
| Fraction unbound (human) (Fu) | 0.268 | 0.268 |
| BBB permeability (log BB) | -0.921 | -0.875 |
| CNS permeability (log PS) | -2.091 | -2.068 |
| Meatabolism | ||
| CYP2D6 substrate | No | No |
| CYP3A4 substrate | Yes | Yes |
| CYP1A2 inhibitor | No | No |
| CYP2C19 inhibitor | No | No |
| CYP2C9 inhibitor | No | No |
| CYP2D6 inhibitor | No | No |
| CYP3A4 inhibitor | No | No |
| Excretion | ||
| Total clearance (log ml/min/kg) | 0.647 | 0.555 |
| Renal OCT2 substrate | No | No |
| Toxicity | ||
| AMES toxicity | No | No |
| Maximum tolerated dose (log mg/kg/day) | 0.429 | 0.430 |
| hERG I inhibitor | No | No |
| hERG II inhibitor | Yes | Yes |
| Oral rat acute toxicity (LD50) (mol/kg) | 2.483 | 2.483 |
| Oral rat chronic toxicity (LOAEL) (log mg/kg-bw/day) | 8.605 | 8.940 |
| Hepatotoxicity | Yes | Yes |
| Skin sensitization | No | No |
| T. Pyriformis toxicity (log |Jg/L) | 0.285 | 0.285 |
| Minnow toxicity (log mM) | 18.400 | 18.952 |
Source: Author’s elaboration.
It is important to note that both cyclopeptides display negative values for the AMES toxicity while the opposite is related to hepatotoxicity. Both peptides will be P-glycoprotein I inhibitors (P-gp I) , being also P-gp substrates. None of the peptides will be inhibitors of the molecules related to cytochrome P450, while the two of them will act as substrates of the CYP3A4 variant. Finally, all the cyclic peptides considered here will display negative results regarding their behavior as hERG I inhibitors and positive with respect to act as hERG II inhibitors. As mentioned earlier, some approximate correlations may be found between the ADMET parameters and the conceptual DFT descriptors. When referring to the absorption properties, it could be seen that the values of the Caco2 permeability, measured as log Papp 10−6 cm/s, correlate directly with the values of the chemical hardness η and inversely with the results for the electrophilicity ω. The VDss (human), measured as log L/kg, correlates in the same way with the conceptual DFT descriptors, while the opposite behavior is found for the BBB and CNS Permeabilities. As the information for the metabolism properties related to the behavior of the variants of the cytochrome P450 are given as Yes or No, it is not possible to establish connections with the values of the conceptual DFT descriptors. For the case of the excretion indicators, the total clearance, expressed as log ml/min/kg, the value for alisporivir is larger than for cyclosporin A, allowing to arrive to a similar conclusion, that is, the values correlate positively with the electrophilicity ω and inversely with the chemical hardness η. An opposite behavior is observed for the oral rat chronic toxicity (LOAEL), expressed as log mg/kg-bw/day, and for the case of the Minnow toxicity (log mM).
Conclusions
As this research is based on the application of a computational methodology developed by our research group for the study of the chemical reactivity of peptides, it is natural to give some impressions about the validation of this methodology for the cases under study. The chemical reactivities of two antimicrobial peptides, alisporivir and cyclosporin A, have been thoroughly investigated by optimizing their structures using the DFTB methodology and calculating their electronic properties using a high-quality model chemistry, namely MN12SX/Def2TZVP/H2O. This model chemistry was already used in previous research, demonstrating its utility for this type of calculations. However, an involved estimation of the KID descriptors for all the molecules demonstrated the ability of the MN12SX density functional for the accurate estimation of the frontier orbital energies based on the KID procedure methodology. The fact that the energy of the LUMO and of the SOMO (or the HOMO energy of the anion) are almost the same, which is reflected in the KID accuracy descriptor ∆SL being very close to zero, is an indication that the derivative discontinuity is negligible for the chosen density functional. This is translated as the ability of the LUMO energy to reflect with precision the electron affinity of the molecule, implying that the chemical reactivity parameters obtained by considering this density functional will be very accurate. This is a very important result because it allowed the estimation of the accuracy of the results based only on the fulfillment of some intrinsic requirements (like the Janak and ionization energy theorems) without the need to resort to the comparison with experimental results that are not be available, as in the present case.
By considering our suggested conceptual DFT-based computational peptidology methodology, both AMPs have been studied by applying certain techniques generally used in the procedure of drug discovery and development. The physicochemical attributes and ADMET (absorption, distribution, metabolism, excretion, and toxicity) indices associated with their bioavailability and pharmacokinetics were forecasted and analyzed as descriptors that could be useful in future drug development research. Although it has not been possible to establish reliable QSAR relationships between the conceptual DFT descriptors and the pharmacokinetics and ADMET indices due to the small number of molecular species studied, it has been possible to find approximate qualitative relationships in terms of their relative values. Thus, we were able to establish some relationships for the Caco2 permeability, the VDss (human), the BBB and CBS permeabilities, the total clearance, the oral rat chronic toxicity (LOAEL) and the Minnow toxicity of alisporivir and cyclosporin A in terms of the chemical hardness η and electrophilicity ω, being directly or inversely. These conclusions pave the way for considering these conceptual DFT reactivity indices as descriptors of the bioactivity in future studies considering a larger number of potential therapeutic drugs.

