











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
En la actualidad gran parte de la producción de combustibles, reactivos químicos para la petroquímica primaria y secundaria, la agricultura o para síntesis de artículos en química fina descansa en el uso de catalizadores, cuya fase activa presenta generalmente tamaños nanométricos. Siendo alguien interesado en la investigación en catálisis o en el uso de nanocatalizadores nos podemos encontrar en cualquiera de estas dos situaciones:
Somos investigadores y hemos encontrado un catalizador maravilloso. Inevitablemente en la carrera académica tendremos que publicar los resultados: ¿cómo convencemos a los demás de que en efecto tenemos un muy buen catalizador?
Trabajamos en una industria y somos los encargados de la producción y, por lo tanto, debemos elegir un catalizador: ¿cómo distinguimos que en efecto el catalizador que nos quieren vender se convierte en una buena inversión?
De las cuestiones anteriores se desprende que la obtención de parámetros cinéticos confiables es fundamental en la investigación, la síntesis de nuevos prototipos de catalizador, creación de nuevos procesos y en la elección de nuevos catalizadores para procesos existentes. La obtención de parámetros cinéticos confiables tiene en realidad dos fases distintas: primero, se realizan mediciones experimentales para obtener una rapidez de reacción (-RA), donde puede considerarse que se obtiene un dato (escalar) por cada medición; y, segundo, a continuación se deben obtener las expresiones matemáticas que se usan para describir la reacción en condiciones diferentes, llamadas la o las expresiones cinéticas.
En este trabajo se presentan las definiciones de catalizador, la rapidez de reacción y también se presenta una descripción sucinta de los reactores de uso común en el laboratorio y se muestran algunos criterios que deberían probarse para que los problemas de transferencia de masa o energía, asociados con el uso de catalizadores porosos para asegurar que los fenómenos de transferencia de masa y energía no interfieran con las mediciones de (-RA) a nivel laboratorio.
¿Qué es un catalizador?
Si queremos crear un proceso, el primer paso es identificar un producto que tenga un alto valor, ya sea porque tiene un valor económico asociado o es un intermediario químico valioso. Cuando estamos interesados en producir algo, es evidente que sería mejor producirlo lo más rápido posible. Pues bien, un catalizador es la sustancia que reduce los tiempos de producción. Una definición actual y simple es: catalizador es una sustancia que aumenta la rapidez con que una reacción alcanza el equilibrio. Otro punto importante es que durante la reacción los sitios activos no se consumen.1 La mayoría de los catalizadores sólidos están constituidos por fases activas de dimensiones nanométricas.
Hablando de producción, se ha introducido al tiempo como una variable importante; así que ahora nos encontramos en los terrenos de la cinética, es decir, de aquellos cambios que ocurren a través del tiempo. Smith y Smith (1981) mencionan que la cinética química se define como el estudio de la rapidez y el mecanismo por medio de los cuales una especie química se transforma en otra. La rapidez de reacción se define como la masa en moles de producto formado o reactivo consumido por unidad de tiempo. El mecanismo es la secuencia de eventos químicos individuales (elementales) cuyo resultado global produce la reacción química observada.
Rapidez de reacción
La función con que vamos a trabajar, de acuerdo con la definición anterior, es la rapidez de reacción (-RA) (Smith y Smith, 1981). La reacción se podría definir como el número de eventos químicos (moles de A que ocurren por unidad de tiempo).
Nótese que son las mismas unidades utilizadas para definir la producción. Así (-RA) es una medida importantísima para describir el desempeño de un catalizador e igual de importante si estamos interesados en la producción en un proceso. Se acostumbra normalizar la rapidez de reacción utilizando la zona donde ocurre la reacción; por ejemplo, si una reacción se presenta en fase homogénea, la reacción ocurre en el volumen de esa fase y entonces la rapidez de reacción se describe como:
En este artículo estamos interesados especialmente en la catálisis heterogénea; por lo que se centrará en catalizadores sólidos; así, la fase de reacción puede ser líquida o gaseosa. En presencia de este tipo de reacciones utilizaremos la masa del catalizador (gramos de catalizador gcat) y entonces:
Conociendo los gcat en la reacción anterior, es posible calcular la cantidad nominal o la carga experimental de fase activa en el catalizador y entonces (-RA) puede expresarse por moles de la fase activa:
A menudo es conveniente normalizar la rapidez de reacción por número de átomos totales expuestos, aquellos con algún número de coordinación específico, o bien, promovidos de manera que la expresión de rapidez contenga información relevante acerca del mecanismo de la reacción. Ahora bien, si tomamos en cuenta que las reacciones catalíticas suceden en los sitios activos, una forma de comparar entre catalizadores es expresando la actividad como el número de moléculas de reactivo convertidas en cada sitio catalítico por unidad de tiempo, a esta medida de la actividad se le conoce como Turnover Frequency (TOF):
Sin embargo, expresar la actividad catalítica por medio de TOF es conveniente únicamente si se conoce inequívocamente la naturaleza del sitio catalítico y se tiene una medida confiable de la cantidad de estos sitios, además de estar en posibilidad de seguir el consumo del reactivo A. De otra forma se puede llegar a consideraciones erróneas o sobre simplificadas acerca de la actividad del catalizador.
Las rapideces de reacción no pueden medirse directamente; se obtienen de la interpretación de los datos de un reactor, donde por cada experimento, se obtiene un valor de (-RA). Ahora nuestro problema es: ¿cómo se podría simplificar la información de varios experimentos para explicar los cambios en (-RA) en otras condiciones de reacción? O, desde otro punto de vista: ¿cómo representamos la rapidez de reacción (-RA)? La mejor opción es una ecuación matemática que nos permita realizar comparaciones entre diferentes catalizadores o hacer predicciones para construir un reactor, a estas expresiones matemáticas se les ha llamado ecuaciones cinéticas.
Con el conocimiento desarrollado en la cinética química e ingeniería de reactores, se ha aceptado que la forma de las ecuaciones cinéticas es:
Donde el estado del sistema se define por: naturaleza de las especies en reacción, composición (concentración, presión), temperatura, presencia de catalizador, naturaleza del contacto entre los reactantes, en reacciones fotocatalíticas, longitud de la radiación incidente, etcétera.
Actualmente, se concibe la rapidez de reacción como una multiplicación de dos funciones de variables independientes (temperatura y composición):
Donde:
f(T) es una función dependiente de la temperatura.
g(composición) es una función dependiente únicamente de la composición.
La función de la temperatura se describe como el coeficiente cinético k (antiguamente constante de velocidad) aquí se incluye la reactividad de las moléculas, que depende de la temperatura y la presencia del catalizador. Con esta información ahora se puede escribir:
En g(composición) se puede utilizar cualquier medida que describa a esta variable. Lo más común es usar la concentración; pero hay otras: por ejemplo, si se tienen gases se puede usar la presión; es común utilizar la conversión (X) y menos común utilizar el avance de reacción (ξ).
Como se habrá notado, se destaca la independencia matemática de ambas funciones. Este comportamiento es muy útil para obtener las estimaciones de los parámetros cinéticos en la rapidez de reacción. La importancia de esta simplificación matemática en las ecuaciones cinéticas radica en que, en virtud de esta independencia, podemos estimar el comportamiento de uno de los parámetros manteniendo constante al otro. Por este motivo, al realizar experimentos para encontrar la rapidez de reacción hacemos experimentos a temperatura constante (isotérmicos) para encontrar la funcionalidad de la concentración, y cuando ya conocemos el comportamiento de la concentración, ajustamos un valor para k. A continuación, trabajando a temperaturas distintas encontramos varios valores de k y de ahí obtenemos la funcionalidad de la temperatura.
Funcionalidad del coeficiente cinético con la temperatura, ecuación de Arrhenius
La función matemática que expresa la relación entre k y la temperatura es la ecuación de Arrhenius:
Donde:
A0 = factor de frecuencia o pre-exponencial. [=] s-1.
EA = energía de activación [=](J)(mol)-1 o (cal)(mol)-1.
R = constante de los gases = 8.3145 (J)(K*mol)-1 o 1.9872 (cal) (K*mol)-1.
T = temperatura [=] K.
Es importante destacar que la temperatura debe expresarse en grados absolutos, también llamados grados Kelvin (K).
Para encontrar los valores de EA y A0 es necesario tener varios valores de k. La ecuación (5) se puede linealizar; permitiéndonos estimar los valores de EA y A0 con relativa facilidad. Al aplicar logaritmos naturales en la ecuación anterior, se obtiene:
Para encontrar los valores de EA y A0 es necesario contar varios valores de k; esto es, después de realizar varios experimentos a diferentes temperaturas y cada uno de ellos en condiciones isotérmicas. Con los valores de k, se construye el grafico ln(k) versus (1/T), que se presenta en la Figura 1. El análisis de la ecuación (6) nos muestra que la ordenada al origen es el valor de ln(A0) y la pendiente de la recta obtenida es igual a EA/T (¡cuidado con eL signo negativo!) Se realiza el ajuste, utilizando, por caso, regresión lineal, del cual obtenemos los valores s de energía de activación y el valor del factor pre-exponencial con cierto grado de confianza estadística.
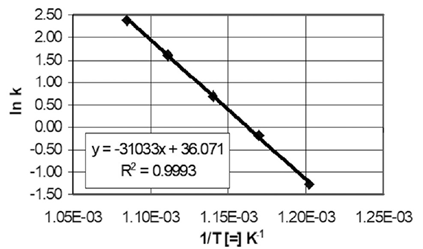
Fuente: Elaboración de los autores.
Figura 1 Ejemplo del ajuste de la ecuación de Arrhenius para una reacción.
Por otra parte, la expresión de f (concentración) puede representarse de varias formas matemáticas que incluyen poca o más información sobre lo que ocurre en la reacción. La más sencilla es la ley de potencias o si somos afortunados encontraremos (o desarrollaremos) expresiones teóricas de rapidez de reacción basadas en el mecanismo de reacción.
Expresiones de la concentración para la rapidez de reacción
Ley de potencias
Desde el siglo antepasado, véase por ejemplo (Berthelot y Saint-Gilles, 1862), se descubrió que existen relaciones simples entre las concentraciones y la rapidez de reacción: por ejemplo, para una reacción dada:
La función de (RA) y la composición se puede representar como:
Donde α, β, ρ y σ, son los llamados órdenes de reacción, se determinan experimentalmente y, por lo tanto, pueden adoptar cualquier valor dentro de los números reales.
Este tipo de expresiones, conocidas como de ley de potencias, se obtienen del ajuste de los datos experimentales; en otras palabras, los órdenes de reacción pueden o no coincidir con los coeficientes estequiométricos. Con esas características, tienen la ventaja de que son fáciles de determinar. Pero tiene varias desventajas, entre ellas: solo son confiables dentro del intervalo donde se obtuvieron los datos experimentales, no se consigue de ellas información acerca de los mecanismos por los cuales se lleva a cabo la reacción misma. En la Tabla 1, encontramos algunas expresiones de rapidez de reacción empíricas.
Tabla 1 Algunas expresiones de rapidez de reacción en función de la concentración (CA) o de la conversión (xA).
| Orden de reacción | En función de CA | En función de xA |
|---|---|---|
| Orden cero | (-RA) = k | (-RA) = k |
| Orden uno | (-RA) = kCA | (-RA) = kCA0(1-xA) |
| Orden dos | (-RA) = kCA2 | (-RA) = kCA02(1-xA)2 |
Fuente: Elaboración de los autores.
Como se muestra en la Tabla 1, se llama orden de reacción al valor del exponente al que está elevada la concentración en la ecuación de rapidez. De este modo, (-RA) = kCA2, donde el exponente de CA es 2 y corresponde a un orden de reacción de orden dos.
Expresiones teóricas o fundamentales de rapidez de reacción
Las expresiones teóricas de velocidad de reacción se basan en la obtención de mecanismos de reacción. Estas expresiones teóricas deben ajustar las expresiones cinéticas obtenidas experimentalmente.
Por ejemplo, la reacción:
tiene como ecuación cinética teórica:
Una ventaja de las expresiones teóricas es que pueden utilizarse en intervalos muy amplios de condiciones experimentales, en contraste con las expresiones de ley de potencias. Por caso, la expresión anterior es capaz de describir cambios en el orden en la reacción; así, partiendo de reactivos puros al inicio de la reacción CHBr~0 tenemos:
Se observa que se presenta orden uno para H2, ½ para Br y cero para HBr.
Por otra parte, cuando la concentración de HBr es grande; por ejemplo, a altas conversiones de Br2 puede despreciarse k2 frente a CHBr /CBr2 y entonces:
Ahora los órdenes de reacción son uno para H2, 3/2 para el Br2 y finalmente negativo para HBr. Una expresión de ley de potencias no puede reflejar cambios en los órdenes de reacción.
Resumiendo, las expresiones de rapidez teóricas o fundamentales están basadas en el mecanismo de reacción. La principal ventaja es que pueden utilizarse en un amplio intervalo de valores y permiten hacer observaciones que mejoran el proceso. La desventaja es que son difíciles de obtener y por lo tanto la disponibilidad de ellas para el diseño de reactores es limitada.
Ecuaciones Langmuir-Hinselwood-Hougen-Watson (LHHW)
Si hablamos de reactores heterogéneos fluido-sólido en donde la reacción se lleva a cabo en la superficie de catalizadores sólidos, la situación es bastante más compleja. Se debe describir una reacción superficial con cantidades medibles; por ejemplo, la concentración (o presión) de los reactivos. Existen diferentes modelos que describen la reacción química superficial; la diferencia principal se refiere a la forma en que se presenta la adsorción o desorción de reactivos o productos. Para la generación de los modelos cinéticos catalíticos, el procedimiento más utilizado es el LHHW2 implementado por Langmuir-Hinselwood-Hougen-Watson. Como su nombre lo indica, este modelo utiliza los conceptos de la isoterma de Langmuir para describir la adsorción.
La suposición principal en que se basa el formalismo LHHW es que el proceso de la reacción química superficial puede dividirse en:
Adsorción de los reactivos, donde se presenta la formación del complejo superficial.
Reacción química superficial, propiamente dicha.
Desorción de los reactivos, que regenera al sitio activo.
El éxito del formalismo LHHW radica en que se establece un procedimiento sistemático que permite la obtención de las ecuaciones. En general, dicho procedimiento se describe de la siguiente manera:
Se elige el tipo de mecanismo que se considera más adecuado.
Se selecciona cuál es la etapa controlante de la reacción.
Se describe la ecuación cinética de ese paso; recordando que es un paso elemental.
Se sustituyen todas las variables que no pueden medirse, utilizando para ello la circunstancia de que las demás reacciones elementales están en equilibrio.
Generalmente, aparece en la ecuación una expresión para los sitios activos, esta se obtiene de un balance de sitios.
Si se considera una reacción:
Las ecuaciones resultantes son del estilo:
Cuando la suposición en el mecanismo es que participan dos sitios adyacentes en la reacción y que la etapa controlante es la reacción superficial. O si se considera otro paso como el controlante:
Donde ahora la suposición es que la etapa controlante es la desorción del producto C.
Si bien las ecuaciones LHHW están ampliamente aceptadas, existen sistemas donde se han reportado diferentes mecanismos con ajustes con una confianza similar; esto ha causado una de las principales objeciones para este tipo de ecuaciones: el número de parámetros en las ecuaciones en ocasiones es demasiado grande para que la elección del mecanismo sea unívoca. En este momento, para que un mecanismo LHHW y la ecuación asociada sean aceptados se requiere comprobar, utilizando técnicas espectroscópicas, que las especies adsorbidas son las requeridas.
Lo que se conoce como ajuste (matemático) de una ecuación cinética es identificar cuáles funciones matemáticas (f(T) y g(concentración)) describen de la mejor manera el comportamiento de la reacción en estudio, y esto incluye asignar los mejores valores a los parámetros en la ecuación (kis o en ecuaciones tipo LHHW, también las KIs ). Dado que las rapideces de reacción no se pueden determinar directamente, debemos utilizar siempre datos experimentales para ajustar la expresión matemática de la reacción cinética. El propósito del ajuste es reducir la diferencia entre el valor de la función matemática supuesta y los resultados experimentales, es decir, el mínimo de la función error. Más información sobre cómo realizar este tipo de ajuste se presentará en la segunda parte de este artículo. Por otra parte, todas las mediciones de rapidez de reacción se realizan en reactores químicos, por eso hablaremos de estos a continuación.
Tipos de reactores experimentales
Para que la catálisis pueda considerarse que sea heterogénea se requiere la presencia de al menos dos fases. Pero por ahora, la mayoría de las reacciones que se aplican a nivel industrial ocurre con un fluido y un sólido; por esta razón, en este artículo de aquí en adelante nos centraremos en la catálisis heterogénea fluido-sólido. Existen varios tipos de reactores que se pueden usar para probar un catalizador sólido, antes de elegir uno u otro reactor es importante decidir qué información queremos obtener.
Para obtener información sobre las propiedades intrínsecas del catalizador, es decir, actividad, selectividad, cinética y desactivación a partir de datos experimentales cualquier tipo de reactor debe cumplir por lo menos con las siguientes características:
Buen contacto entre los reactantes y el catalizador.
Ausencia de limitaciones de transporte de masa y energía, entre las partículas de nanocatalizador y dentro de las mismas.
Distribución de tiempos de residencia conocidos bajo condiciones isotérmicas.
Dos tipos de reactores usualmente utilizados que cumplen con los requisitos mencionados anteriormente, son el reactor de flujo pistón (PFR) y el reactor de tanque agitado de flujo continuo (CSTR). Generalmente, estos reactores son operados en estado estacionario para disminuir los costos y complejidad de la operación. El reactor intermitente, también llamado, por lotes o batch es bastante común a nivel laboratorio, con él además se obtienen resultados relacionados directamente con el tiempo. Otros tipos de reactores son operados en estado transitorio, por ejemplo el de pulsos (temporal analysis of products) (Gleaves, Ebner y Kuechler, 1988). Estos últimos reactores tienen una operación más compleja y se usan principalmente para establecer mecanismos de reacción.
Reactores experimentales que operan en estado estacionario
Reactores de lecho fijo y flujo pistón
Otro de los reactores de amplio uso en el laboratorio es el reactor de lecho empacado (packet bed reactor: PBR) o reactor de lecho fijo (fixed bed reactor: FBR). Con diseño apropiado, el reactor FBR se adecua bastante bien al modelo de reactor de flujo pistón, consiste, básicamente, en un tubo que puede construirse con materiales diversos y está empacado con un catalizador sólido. El reactivo se mueve a través del catalizador en flujo turbulento. A nivel laboratorio se busca conseguir un perfil sin variación de las velocidades de flujo, lo que garantizaría la ausencia de gradientes radiales de temperatura y concentración (Perego y Peratello, 1999). Un reactor PFR experimental se muestra en la Figura 2.
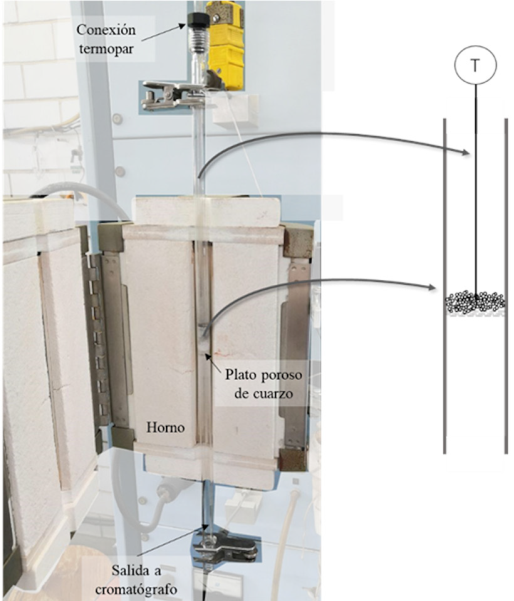
Por lo general, existen dos modos para operar los reactores empacados. En el primer modo de operación denominado “diferencial” la conversión de reactivo a la salida del lecho catalítico debería ser menor al 5% (Fogler, 1992). Esta condición se utiliza buscando que las variaciones de la concentración a la entrada y salida sean proporcionales al cambio en la conversión. Por otro lado, un reactor operado en modo ‘integral’ es aquel en el cual la conversión excede el 5%.
Reactores de tanque agitado continuo
Estos reactores también conocidos como reactores CSTR (por sus siglas en inglés: continuous stirred tank reactor) consisten en tanques en los cuales se asegura que no existen gradientes de temperatura o concentración resultado del mezclado de los reactivos en el tanque. Cuando estos reactores están operando de forma muy cercana a la ideal. Usando estos reactores es posible obtener la rapidez de reacción usando una simple expresión algebraica expresada en términos de la concentración a la salida del reactor.
En los reactores CSTR que utilizan catalizadores sólidos, se debe considerar que las partículas de catalizador deben estar suspendidas de forma homogénea. Esto no siempre es posible debido a la diferencia de densidades entre sólidos y fluido, especialmente en el caso de suspensiones líquidas. Para asegurar el adecuado contacto entre los reactivos y las partículas catalíticas dentro de este tipo de reactores se construyeron los llamados reactores de canasta. En estos reactores, el catalizador se encuentra retenido en una malla fina que permite el paso del líquido e impide la salida de catalizador. Existen dos configuraciones: en la primera, el catalizador se encuentra en una canastilla estática y una combinación de agitadores y bafles dirigen el flujo hacía la canastilla para asegurar un contacto adecuado entre reactivos y catalizador; como segunda opción, la canastilla que contiene al catalizador se une al agitador, esta configuración en conjunto con bafles fijos asegura el contacto adecuado del catalizador con los reactivos (Pergo y Peratello, 1999; Carberry, 1964).
Reactores experimentales que operan en estado transitorio
Reactor por lotes (intermitente o batch)
Estos reactores consisten en un recipiente con agitación vigorosa; por tanto, se espera que el catalizador y los reactivos presenten un buen contacto. El experimento se lleva a cabo al operar durante un tiempo determinado a una presión y temperatura determinados. El progreso de la reacción es monitoreado periódicamente analizando el contenido del reactor, para lo cual se deben extraer alícuotas que no alteren significativamente el volumen del reactor.
De forma experimental, el catalizador se puede colocar libre o bien en una canastilla, como las mencionadas anteriormente para el reactor CSTR, dependiendo del tamaño de partícula y densidad del fluido.
Una configuración común para las reacciones gas-líquido-sólido son los reactores semi-batch, en los cuales uno de los reactivos se alimenta continuamente al reactor de modo que su concentración permanezca controlada durante el desarrollo de la reacción.
Reactores de pulsos
En este tipo de reactores, después de establecer una operación estable se hace una perturbación en las condiciones de reacción y se miden los cambios que presente la reacción en función del tiempo para obtener información cinética. En la práctica, las perturbaciones en la concentración son las más comunes y, por lo general, se inyecta una cantidad pequeña y conocida de reactivos en la alimentación que pasa a través del lecho y se monitorean los cambios en la conversión a la salida del lecho (Pérez-Ramírez et al., 2000).
Criterios para evaluar problemas de transferencia de masa y energía en reactores
Los sistemas de reacción catalíticos heterogéneos fluido-sólido son sistemas complejos desde el punto de vista de la transferencia de masa y energía; pues están constituidos por diversas fases, además de que se presentan fenómenos a diversas escalas. Por ejemplo, los reactores de hidrogenación de hidrocarburos insaturados utilizan por lo general catalizadores que contienen nanopartículas de algún metal como paladio o platino (Siegel, 1978). En estos reactores podemos identificar que los reactivos (hidrocarburos insaturados e hidrógeno) deben desplazarse hacia el interior de los poros de las partículas catalíticas para acceder a los sitios activos donde se lleva a cabo la reacción, de forma esquemática esto se muestra en la Figura 3.

Fuente: Elaboración de los autores.
Figura 3 Representación esquemática de un reactor empacado y un catalizador sólido impregnado con un metal.
Nos detendremos un poco más en la transferencia de masa externa e interna. La situación que se presenta es que el pellet del catalizador está fijo en el espacio y el fluido golpea la partícula. Los fenómenos de transporte enseñan que se forma una capa límite alrededor del pellet, donde no hay movimiento convectivo. En cada pellet, también se generan perfiles mostrados esquemáticamente en la Figura 4. Cuando sintetizamos un catalizador buscamos que la fase activa esté bien dispersa dentro del sistema poroso del catalizador. En el sitio catalítico ocurre la reacción, por caso: A → B. La causa de la transferencia de masa dentro del catalizador (o transferencia de masa interna o intraparticular) es la reacción misma. En el sitio activo desaparece A (se forma B) y la concentración de A disminuye; con la diferencia de concentración, A se difunde de una ubicación con mayor concentración a la de menor concentración. Dado que A en su recorrido hacia el centro de la partícula probablemente se encuentra con los sitios catalíticos, por eso en el centro de la partícula la concentración tiene un mínimo y A debe transferirse desde fuera del catalizador hacia adentro del mismo. De manera simultánea, cada mol de A que reacciona genera calor (exotérmica) y entonces la partícula se calienta y ocurre la transferencia de calor; es necesario tomar en cuenta este efecto en el coeficiente cinético. En la situación en que se trabaja una reacción endotérmica la partícula se enfría. En las mediciones de los datos de (-RA) se debe evitar que el perfil de concentraciones caiga demasiado y en cambio, que el de temperatura preferentemente sería casi isotérmico.

Fuente: Elaboración de los autores.
Figura 4 Transferencia de masa y energía interna en un catalizador.
Finalmente, en la parte externa de la partícula no hay reacción, pero dado que se consume A en el catalizador, la concentración en la boca de los poros es menor que en el seno del fluido y tenemos transferencia de masa desde afuera del catalizador (transferencia de masa externa o interparticular). El problema extremo que debemos evitar es el marcado en rojo en la Figura 4. Si existen graves problemas en la transferencia de masa externa, la concentración dentro del catalizador puede ser muy baja y se reportaría una velocidad casi nula, lo cual sería un error. Resumiendo, los reactivos deben transportarse desde el seno del fluido hasta la superficie externa de la partícula catalítica, este fenómeno se presenta a través de la capa límite y se conoce como transferencia de masa externa o transferencia de masa interparticular. Una vez que los reactivos se encuentran en la superficie de la partícula catalítica, los reactivos se transportan a través del sistema poroso del catalizador hasta el sitio activo, este fenómeno se conoce como transferencia de masa intraparticular o transferencia de masa interna. El reactivo A se adsorbe en el sitio catalítico y reacciona. Una vez que ocurre la reacción, los productos de reacción se deben desorber desde el sitio activo donde ocurrió la reacción, a través de los poros de la partícula catalítica hasta el seno del fluido. Existen los pasos equivalentes para el transporte de energía.
Así, tenemos que debido a la presencia de dos fases (reactor heterogéneo) se generan gradientes de masa y energía, los cuales son inevitables en la práctica industrial. Debido a que en el nivel laboratorio se está interesado en determinar la rapidez de reacción tan solo del fenómeno químico debemos asegurar que los fenómenos de transferencia de masa y energía no alteran nuestras mediciones. Los criterios siguientes, se refieren a las limitaciones a la transferencia de masa o energía. Para obtener datos representativos de la cinética de la reacción es necesario realizar, previamente a los experimentos a reportar o considerados válidos, las comprobaciones experimentales y numéricas enumeradas a continuación para decidir si la presencia de estos problemas es tan pequeña que confiar en nuestras mediciones es posible. Empezaremos con los problemas a escala macro en el reactor y después en la escala micro en el pellet catalítico.
Criterios para evaluar problemas de transferencia de masa y energía en reactores de flujo pistón (PFR) y reactores de lecho empacado3
En la práctica experimental, los reactores de lecho fijo (FBR o PBR) son muy utilizados a nivel laboratorio para evaluar catalizadores sólidos con reactivos y productos en fase gaseosa o líquida. Una vez decidido el tipo de reactor más adecuado para obtener la información necesaria, es conveniente tomar algunas precauciones que aseguren que el comportamiento del reactor es lo más cercano al comportamiento ideal. Resulta que bajo condiciones adecuadas (ver abajo) el modelo matemático más simple de un PBR es muy similar, matemáticamente, al del reactor PFR. Y esto simplifica enormemente la evaluación de (-RA).
Independientemente del tipo de reactor, cuando se realizan mediciones de rapidez de reacción para un sistema catalítico fluido-sólido, podríamos encontrar dos definiciones de rapidez de reacción: La rapidez de reacción intrínseca (en la que en realidad estamos interesados) que se refriere a los cambios causados solamente por la reacción química y la reacción global que incluye los fenómenos químicos y los de transferencia de masa y es la que la naturaleza nos proporciona. Así, la clave es cómo asegurar que el valor de (-RA) medido sea lo más cercano posible a la velocidad intrínseca. Dicho de otra manera, si podemos probar que las mediciones se realizan sin problemas de transferencia de masa y energía, el procedimiento para obtener (-RA) es directo y simple. En caso contrario, debemos estimar la magnitud de cada efecto y separarlo de la medición de la velocidad de reacción. Por lo tanto, a continuación, se muestran los criterios para asegurar mediciones adecuadas de la actividad y selectividad intrínseca del catalizador utilizando un reactor de flujo pistón (PBR).
Criterio para despreciar el efecto de la dispersión axial en el reactor de lecho empacado
Una de las características más importantes de un reactor de flujo pistón (de hecho, aquella característica que le da nombre) es que el tiempo de residencia dentro del rector de todos los elementos de fluido es el mismo. Como consecuencia, el flujo tiene un perfil de velocidades plano a lo largo del reactor. La difusión radial, axial y entre partículas, así como los efectos de pared pueden causar desviaciones del flujo ideal. Se considera que, si el tamaño de las partículas es suficientemente pequeño respecto a la longitud de la cama, la difusión radial será pequeña. Así pues, uno de los primeros criterios se basa en la relación entre la longitud de la cama (Lb) y el diámetro de la partícula, es el siguiente:
Si este criterio se cumple, tendríamos desviaciones menores al 5% consideradas como no causantes de cambios importantes en el patrón de flujo.
Criterio para no considerar los efectos asociados a la pared del reactor
En los reactores de lecho fijo la uniformidad del empaque de la cama cambia cuando nos acercamos a la pared del reactor, además, las superficies de las paredes del reactor se pueden considerar planas (comparadas con las partículas). Esto provoca que el flujo cercano a las paredes se puede considerar más rápido que la rapidez de flujo promedio a esa altura del lecho. Para los gases, el efecto de la pared puede ser ignorado si la relación entre el diámetro del lecho empacado y el diámetro de la partícula es mayor a 10.
Si este criterio se cumple, entonces podemos considerar que el perfil de velocidades dentro del reactor se asemeja a flujo pistón.
Por otro lado, en las reacciones líquido-sólido, debido a que en líquidos se tiene una difusividad menor se recomienda usar un diámetro de lecho máximo, cuyo valor se calcula con la ecuación 16.
Criterio para no preocuparse por la caída de presión en el lecho de catalizador
La ecuación que se utiliza para calcular la caída de presión en el catalizador es conocida también como ecuación de Ergun (Ergun, 1952). Una regla de dedo (empírica) para reactores indica que la caída de presión debe ser menor al 20% de la presión total de operación.
Criterio para ignorar el gradiente de temperatura radial dentro del reactor
Cuando se realiza una reacción se genera o consume calor dependiendo de si la reacción es exotérmica (o endotérmica). A la entrada del reactor la reacción ocurre más rápido, por lo que la generación (o consumo) de calor es mayor cerca de la entrada del reactor. De manera similar, se puede concluir que en el centro del reactor se alcanzan temperaturas más altas. Esas diferencias de temperatura crean diferencias en las rapideces de reacción que estamos midiendo. El siguiente es el criterio que sirve para decir que es posible ignorar el efecto del gradiente de temperatura radial en el reactor.
Una vez que se realizan las comprobaciones numéricas con los criterios anteriormente mencionados, se deben realizar pruebas experimentales.
Para establecer los gradientes de temperatura entre partículas se deberá variar el flujo y la cantidad de catalizador manteniendo el espacio velocidad, W/FA0 constante, si no hay problemas de transferencia de masa la conversión en estos experimentos debe ser similar. Es importante considerar que la existencia de puntos calientes dentro del reactor puede compensar algunos problemas de transferencia de masa, por lo tanto, se debe asegurar una dilución adecuada de las partículas catalíticas especialmente con materiales de baja conductividad térmica.
Para corroborar experimentalmente la ausencia de gradientes de concentración intraparticulares se deberá variar el tamaño de partícula, y realizar la reacción manteniendo los otros parámetros constantes (T, relación W/FA0, gasto másico). Si la conversión se mantiene igual en estos experimentos, se puede decir que la reacción está controlada cinéticamente y es independiente del tamaño de partícula.
La dilución del lecho catalítico con materiales químicamente inertes y de baja área superficial como cuarzo, a-alúmina, vidrio, y carburo de silicio (SiC), se usa ampliamente al mejorar la conducción de calor dentro del lecho catalítico y disminuir la caída de presión. Sin embargo, debe tomarse en cuenta que una alta dilución puede causar heterogeneidades en la distribución del catalizador, así como ‘caminos preferenciales’ para el flujo dentro del reactor debido a la diferencia de densidades entre estos materiales y las partículas catalíticas (Pérez-Ramírez et al., 2000).
Por lo tanto, es conveniente limitar la dilución de la cama en función del tamaño de partícula catalítica y la longitud del lecho para limitar las desviaciones de la idealidad al 5%.
Criterios relacionados con el pellet del catalizador
Criterio para no tomar en cuenta las limitaciones a la transferencia de masa entre partículas (transferencia de masa externa)
El criterio que se utiliza se basa en el número de Carberry (Ca). En este caso si Ca <0.05/(|n|) resulta que se puede ignorar la transferencia de masa externa. Utilizando la definición del Ca se tiene la siguiente ecuación:
Criterio para prescindir de las limitaciones a la transferencia de masa intraparticular
Si se presentan problemas de transferencia de masa dentro de la partícula (o intraparticulares) significa que el transporte de masa de los reactivos dentro de la partícula es deficiente. Si los reactivos no se difunden, entonces no llegan al sitio catalítico y no aprovechamos todo el potencial del catalizador. Esta medición se realiza a través del factor de efectividad η, si η<<1 estaríamos midiendo una rapidez de reacción distinta a la real, es decir, estaríamos reportando una medida errónea. Este criterio, para una reacción de orden ‘n’ en un reactor isotérmico, utiliza el módulo de Wheeler-Weisz (Weisz y Prater, 1954). Empleando el análisis de Mears, se puede decir que si el número adimensional Φ = η ϕ2 (Φ es otro número adimensional resultado de la multiplicación del factor de efectividad (η) por el módulo de Thiele (ϕ) es menor a 0.15, entonces podemos considerar que las limitaciones de transferencia de masa son despreciables.
Criterio para despreciar las limitaciones a la transferencia de calor entre partículas
La ecuación de Arrhenius señala que con el aumento de temperatura el valor del coeficiente cinético se incrementa exponencialmente. Por otro lado, toda reacción tiene un calor de reacción asociado. Si existen problemas de transferencia de calor entre partículas y la reacción es exotérmica, el calor generado provoca un aumento de la temperatura local. Esto es, se calienta la partícula catalítica y entonces este calor se transfiere a los alrededores. Si la reacción es endotérmica, la temperatura disminuye. Si no se consideran estos efectos, es evidente que en ambos casos se está midiendo una rapidez de reacción equivocada. El criterio para decir que no existen problemas de transferencia de calor entre partículas son:
En general, una reacción muy exotérmica o endotérmica en combinación con una conductividad térmica baja en el lecho catalítico puede causar gradientes de temperatura dentro de las partículas catalíticas y entre ellas, en estos casos es recomendable diluir el lecho con algún material inerte.
Criterio para menospreciar las limitaciones a la transferencia de calor intraparticular
Si la partícula se calienta (o enfría, si la reacción es endotérmica) en demasía debido al calor de reacción, las mediciones de la rapidez de reacción no serán correctas. En esta ocasión también encontramos a Φ y al número de Arrhenius γ, en la definición del criterio:
Ejemplos de medidas de actividad catalítica en la literatura
Si bien en los apartados anteriores expresamos las ecuaciones de rapidez de reacción en función de la concentración, temperatura, y se presentaron los criterios para posibilitar minimizar los problemas asociados con la transferencia de masa y energía cuando realizamos determinaciones de (-RA); en la literatura científica encontramos una amplia variedad de formas en las cuales se reportan ecuaciones de rapidez de reacción, desde aquellas reacciones “modelo” en las cuales se busca encontrar el mecanismo cinético y se requiere observar incluso los reactivos adsorbidos, hasta reacciones más complejas en las cuales se utiliza un sistema de reacciones simplificado, y únicamente se cuantifica la conversión. Incluso podemos encontrar reportes en los que, aparentemente, ignorarían los temas antes mencionados, pero con poderosas razones para operar de esa manera. A continuación se exponen algunos ejemplos, comenzando con una reacción sencilla y terminando con reacciones más complejas. En el artículo “Obtención y análisis de expresiones de cinética química. II. Optimización y evaluación de parámetros cinéticos” (ver en este número) se encontrarán otros ejemplos.
Reacción de oxidación de CO
Empezaremos con un ejemplo relativamente simple. La reacción de oxidación de CO + O2 → CO2. Esta reacción de oxidación se ha estudiado en catalizadores de Au soportado en titania o ceria y ha sido una de las reacciones más estudiadas. A pesar de la sencillez de la ecuación estequiométrica, actualmente se han propuesto múltiples mecanismos y su correspondiente ecuación cinética teórica, pero no se ha alcanzado consenso acerca de la naturaleza de los sitios activos y los detalles del mecanismo. Varios investigadores han desarrollado técnicas que permiten “observar” las especies adsorbidas y conocer su concentración.
El trabajo de Del Río (2014), por caso, mostró que el CO se adsorbe tanto en especies de Au0 como Auδ- y que mientras las especies adsorbidas en Auδ- son rápidamente oxidadas a CO2, el CO adsorbido en Au0 se transforman más lentamente; asimismo, se encontró que la presencia de CO2 en la fase gas retarda la oxidación de las especies de CO adsorbido en Au debido a la adsorción competitiva entre CO y CO2 en vacantes de oxígeno.
En este caso se usó un microrreactor de pulsos y técnicas espectroscópicas, lo que permitió expresar el mecanismo en función de las especies adsorbidas sobre distintos sitios de oro en el catalizador. Específicamente para la oxidación de CO adsorbido en sitios de Auδ- se pudo plantear la siguiente ecuación:
Donde se observa que la expresión incluye la especie CO adsorbida sobre Au, lo que no puede expresarse con una ley de potencia.
Producción de biocrudo utilizando microalgas
El biocrudo es un combustible cuyo origen es la biomasa y tiene una composición química similar a la del petróleo crudo de origen fósil. Debido a que durante la descomposición pirolítica en presencia de un catalizador la biomasa produce un número muy grande de compuestos a partir de varios tipos de biocompuestos (proteínas, carbohidratos y azúcares), en esta reacción se desconocen las posibles rutas de reacción. Esto impide que se pueda seguir la reacción con las variables “típicas” de conversión (X) o incluso de concentración. Una práctica común en la literatura es reportar los balances de masa y rendimientos en masa, como se muestra en la Figura 5.
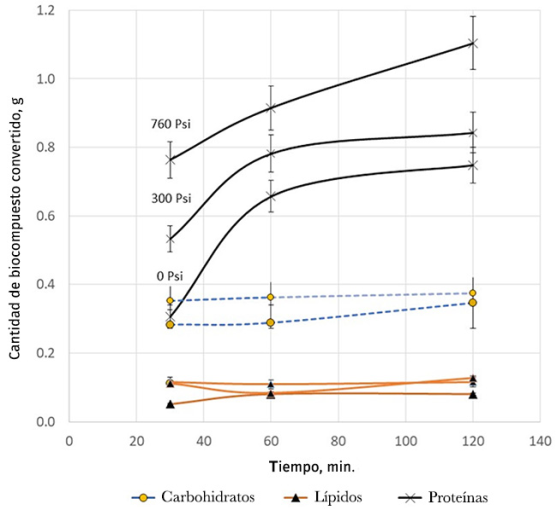
Fuente: Elaboración de los autores.
Figura 5 Conversión por biomolécula en el proceso de solvólisis de un consorcio de microalgas rico en Spirulina sp utilizando zeolita HZSM-5 en función de la presión.
Utilizando el experimento anterior, una primera aproximación de las ecuaciones de rapidez de reacción podría ser:
Las ecuaciones anteriores suponen que cada familia de bio-moléculas reacciona de forma irreversible y que todas las reacciones pueden considerarse inicialmente de orden 1. Pero proceder de esta manera no nos proporciona información sobre qué tipo de productos se están obteniendo.
Conclusiones
En la obtención de expresiones cinéticas para la descripción de una reacción es necesario partir de mediciones experimentales de la rapidez de reacción. La expresión cinética es una función matemática que nos permite comparar entre varios catalizadores, diseñar reactores y estudiar la influencia de las variables de operación, entre otras cosas. Por lo cual es imprescindible que la ecuación represente adecuadamente los resultados experimentales y que estos, a su vez, proporcionen información de la reacción catalítica. Cuando se está trabajando con catalizadores sólidos es necesario evitar los problemas de transferencia de masa y energía. De tal modo, en este artículo se dieron varios criterios que permiten asegurar que las mediciones de rapidez de reacción únicamente consideran la reacción en sí misma, esto nos posibilita obtener datos correctos acerca de la actividad de nuestros catalizadores.

