











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkPresentamos el caso clínico de un paciente de 45 años, con antecedentes de tabaquismo activo de más de 20 años de evolución. El paciente consultó por síndrome constitucional, asociado a tos seca y disnea de moderados esfuerzos. En la radiografía de tórax se objetivó una consolidación en el lóbulo inferior derecho. El estudio se completó con tomografía computarizada (TC) toracoabdominal y tomografía por emisión de positrones, que confirmó la presencia de una lesión de 17 cm que ocupaba la totalidad del lóbulo inferior derecho con invasión de la grasa paracardiaca y derrame pleural adyacente (Fig. 1), siendo sugestivo radiológicamente de tumor fibroso solitario pleural (TFSP). Se realizó una biopsia con aguja gruesa en la que se obtuvieron cuatro cilindros que mostraron una tumoración fusiforme moderadamente celular con un estroma esclerótico. Las células que la componían eran de mediano tamaño, con citoplasma mal definido y núcleos ovalados vesiculosos, sin intensa anisocariosis ni nucléolo prominente. Se determinó la presencia de una mitosis en cinco campos, y no se observó necrosis en los cilindros estudiados. La inmunohistoquímica mostró expresión de CD99, BCL-2 y CD34, siendo compatibles estos hallazgos con el diagnóstico de TFSP.

Figura 1 Fotografía macroscópica al corte en la que se muestra la lesión tumoral En el lado izquierdo se observa un corte de la lesión donde se ve su crecimiento difuso y la destrucción del parénquima. En el lado derecho se observa otra zona de la lesión mejor delimitada y respetando el parénquima circundante.
El caso se presentó en el comité de tumores para decidir el mejor manejo multidisciplinario de la lesión, dado su gran tamaño, y se decidió resecarla mediante lobectomía. En un primer momento se realizó una exploración mediante toracoscopia derecha bajo anestesia local y sedación profunda, con hallazgo de una gran masa con aspecto intraparenquimatoso. La exploración fue limitada por el gran volumen de la tumoración, que se encontraba adherida a la pleura parietal. En un segundo tiempo se realizó una toracotomía posterolateral derecha con acceso por doble espacio intercostal (quinto y octavo), y a través de ella, una lobectomía inferior derecha con linfadenectomía hiliomediastínica sistemática. La cirugía se realizó sin incidencias importantes y el posoperatorio transcurrió con normalidad.
En la descripción macroscópica de la pieza quirúrgica se destacó la presencia de una gran tumoración de aspecto de sólido, de color blanquecino amarillento con pequeñas zonas quísticas y de necrosis, y focos hemorrágicos (Fig. 1). El tamaño de la lesión era de 17,5 × 16 × 9 cm e infiltraba el parénquima pulmonar adyacente afectando el margen de resección (R1). Desde el punto de vista microscópico, destacaba la gran heterogeneidad tumoral; por un lado, había zonas con intensa celularidad y morfología de célula pequeña, y por otro lado, zonas acelulares formadas por fascículos irregulares de núcleos ovales separados por bandas de colágeno. Asimismo, llamaban la atención las numerosas figuras de mitosis (15 mitosis por 10 campos de gran aumento) y las abundantes zonas de apoptosis y áreas de necrosis tumoral (Fig. 2), lo que contrastaba con la muestra de la biopsia previa. El índice de proliferación celular (Ki 67) fue del 80% en algunas zonas1-3. Toda la inmunohistoquímica realizada sobre la pieza tumoral fue negativa: marcadores mesoteliales (calretinina, D2-40, WT-1), epiteliales (AE1/AE3, CK19, CK7, CK5/6, EMA), neuroendocrinos (cromogranina, sinaptofisina, CD56), melánicos (Melan A, S-100) y musculares (actina, desmina). Asimismo, la translocación del sarcoma sinovial también resultó negativa. Dado que en nuestro hospital en ese momento no se disponía de STAT-6, no se pudo determinar este marcador, que habría aportado un valor diagnóstico importante dadas sus altas sensibilidad y especificidad para el TFSP. Tras valorar todas las opciones y como diagnóstico de exclusión, se concluyó que la pieza era un TFSP de características malignas.

Figura 2 A: fotografía microscópica (H-E) de las zonas hipercelulares. B: numerosas figuras de mitosis. C: áreas más hipocelulares de morfología más convencional de TFSP.
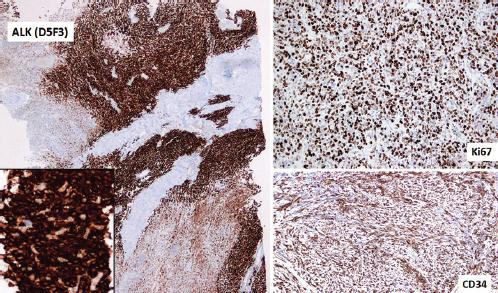
Al poco tiempo de la intervención, el paciente comenzó con dolor abdominal y elevación de las transaminasas, por lo que se solicitó una TC que mostró lesiones ocupantes de espacio hepáticas de nueva aparición, confirmándose mediante punción con aguja fina su origen metastásico. Debido a lo inusual del caso, en el que nos encontramos ante un TFSP con datos de malignidad y progresión sistémica precoz tras la intervención, para el cual no existe un estándar de tratamiento sistémico, se decidió ampliar con un panel mutacional, con el objetivo de encontrar una posible alternativa a la quimioterapia sistémica en una patología infrecuente y actualmente huérfana de tratamiento3. Se estudiaron EGFR, ROS 1, c-Kit y ALK, y se encontró expresión de la proteína ALK mediante inmunohistoquímica (Fig. 3). Este hecho se confirmó posteriormente mediante la técnica de hibridación in situ fluorescente, por lo que se planteó el inicio de tratamiento sistémico con crizotinib. Sin embargo, dado el mal estado general del paciente no fue posible, y este falleció poco después a causa de un fallo hepático.
Discusión
El TFSP es un tumor muy infrecuente que fue descrito por primera vez en 19311. Puede presentarse en diferentes localizaciones, incluyendo tejidos blandos y órganos sólidos, pero lo más habitual es que se origine en la cavidad pleural, representando menos del 5% de todas las neoplasias torácicas. Su etiopatogenia no esta del todo clara, aunque parece probable que tenga un origen mesenquimal. En cuanto a su presentación clínica, lo más habitual es que aparezca entre la quinta y la sexta décadas de la vida, lo cual difiere de nuestro caso, que tuvo una presentación más precoz. Además, no parece haber diferencias por sexo ni claros factores de riesgo identificados2,3.
Debido a que se trata de un tumor muy infrecuente, su comportamiento clínico no se conoce por completo. En general se trata de un tumor benigno de crecimiento lento, con síntomas compresivos, como disnea, tos o dolor torácico, como en nuestro paciente. De hecho, por su comportamiento indolente y poco agresivo, suele ser habitual su diagnóstico incidental al realizar una radiografía de tórax por otro motivo4. Por ello, lo más frecuente es que al manifestarse se trate de una enfermedad en estadio localizado, con una masa definida frecuentemente pediculada, de forma que su exéresis puede ser completa. Lo más habitual es que se intervenga mediante videotoracoscopia, con verificación intraoperatoria de márgenes de resección libres. Sin embargo, cuando el tumor se presenta como una gran masa (como en nuestro caso clínico), el abordaje por toracoscopia no es posible y se utilizan otros más radicales, como la toracotomía ampliada. No obstante, la técnica quirúrgica no parece influir en el pronóstico. El único factor que se ha relacionado con un menor riesgo de recaída es la extirpación total de la masa, siendo fundamental dejar unos buenos márgenes de resección.
Las diferentes series comunicadas estiman el riesgo de recaída tras la resección de la lesión en torno al 10-25%, con una supervivencia media a los 10 años del 73-100%5,6. El motivo por el cual un TFSP presenta un comportamiento más agresivo de lo esperado, como en nuestro caso, no se conoce. Como factores a analizar es importante determinar en la pieza histológica si existen criterios de alta agresividad: presencia de necrosis, pleomorfismo nuclear, infiltración de tejidos adyacentes (pulmón, partes blandas de la pared torácica), áreas de desdiferenciación y elevado índice mitótico (más de tres mitosis en 10 campos de gran aumento)7. Estos datos, junto con la presencia de cirugía incompleta, enfermedad metastásica al debut y tumor > 10 cm máximo, orientan hacia la presencia de un tumor más agresivo y de peor pronóstico. Se ha publicado un estudio retrospectivo que plantea la posibilidad de una escala con valor predictivo en la recurrencia de este tumor. Dicha escala se ha validado en una muestra de 113 pacientes, en los que se evaluaron el origen pleural o no de la lesión, la morfología, el tamaño, la presencia de necrosis o hemorragia, y el número de mitosis, siendo clasificados los pacientes en bajo o alto riesgo de recurrencia. El tamaño > 10 cm, la hipercelularidad, la necrosis o la hemorragia, y cuatro mitosis por campo se relacionaron con un aumento de la recidiva a largo plazo8. Además, este último se identificó como factor independiente de recidiva a largo plazo. Según esta escala, nuestro paciente presentaba un alto riesgo de recaída, lo que se tradujo en una evolución tórpida.
Respecto al tratamiento sistémico del TFSP existen pocos datos, debido a su baja frecuencia y a su comportamiento mayoritariamente benigno. Cuando nuestro paciente progresó de forma precoz tras la cirugía, nos planteamos cuál podría ser la mejor opción terapéutica. En cuanto a la posibilidad de quimioterapia, tan solo existen datos de series retrospectivas en las que se ha evaluado el posible papel de las antraciclinas, la ifosfamida, los platinos y los taxanos. Todos estos estudios muestran unas tasas de respuesta pobres, con una supervivencia libre de progresión de unos 4 meses y con una toxicidad importante9-11. También se barajó la posibilidad de iniciar tratamiento con inhibidores de la tirosina cinasa, por presentar mejor perfil de toxicidad, con medianas de supervivencia similares a las de los tratamientos citotóxicos12,13.
Sin embargo, valorando las escasas opciones terapéuticas disponibles, y dado que existía la posibilidad en nuestro centro de realizar un panel mutacional con el objetivo de encontrar una diana terapéutica en el marco de la medicina personalizada, optamos por esta última opción y encontramos una translocación de ALK, y con ello la posibilidad de un tratamiento eficaz. Como es conocido, esta alteración se usa como diana terapéutica en diferentes tipos tumorales, y existen diferentes fármacos altamente eficaces que han cambiado el pronóstico y la evolución de estas neoplasias. Lo más frecuente es que se encuentre asociada al cáncer de pulmón no microcítico, pero en la literatura se ha descrito la presencia de dicha translocación en ciertos tumores raros, como el histiocitoma fibroso epitelioide y algunos sarcomas de partes blandas poco frecuentes14. Cuando se identifica esta translocación se debe iniciar el tratamiento dirigido con inhibidores específicos, como crizotinib, ceritinib o alectinib, obteniendo una rápida respuesta y medianas de supervivencia que pueden alcanzar los 20 meses15. En nuestro caso clínico, a pesar de identificar esta diana no se pudo iniciar el tratamiento idóneo debido al rápido deterioro del paciente y su fallecimiento poco después.
Este hecho hace que reflexionemos acerca de si, en la era de la medicina personalizada, tumores raros como el TFSP, de los que se desconoce su evolución clínica y se carece de tratamientos eficaces, no se deberían encuadrar dentro de las enfermedades raras y con necesidad de una buena tipificación mediante técnicas de biología molecular, con el objetivo de un tratamiento personalizado.

