











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
El carcinoma de células de Merkel (CCM) es un carcinoma cutáneo con diferenciación neuroendocrina con una incidencia en aumento (estimada en 0.7/100,000 personas/año). Su patogénesis está relacionada con el poliomavirus asociado al carcinoma de Merkel (MCPyV, Merkel cell polyomavirus) y con la exposición crónica a la radiación ultravioleta (RUV)1. Clínicamente suele presentarse como un nódulo único eritematoso de rápido crecimiento y asintomático en regiones fotoexpuestas de personas de edad avanzada con piel clara, a menudo simulando lesiones benignas. Se caracteriza por su alta recurrencia local y la presencia de metástasis ganglionares, lo que disminuye la supervivencia a los 5 años al 35%, o al 14% si existen metástasis a distancia2-4. Los avances en el conocimiento de la fisiopatología y la genética molecular de esta enfermedad han permitido ampliar el arsenal terapéutico; sin embargo, la cirugía sigue siendo de primera elección.
Método
El objetivo del presente artículo es realizar una revisión bibliográfica de lo publicado hasta la fecha acerca del CCM, sus características clínicas y anatomopatológicas, y su manejo terapéutico a raíz de los casos diagnosticados en nuestro centro en el período de enero de 2018 a enero de 2019 (n = 2), con el fin de profundizar los conocimientos de esta afección tan poco frecuente y evaluar el manejo diagnóstico-terapéutico que se ha llevado a cabo. Realizamos una búsqueda bibliográfica en PubMed con las palabras clave Merkel Cell Carcinoma y aparecieron 3308 artículos, de los que seleccionamos 10 para lectura completa y 22 para lectura del resumen acorde con su contenido y fecha de publicación.
Caso 1
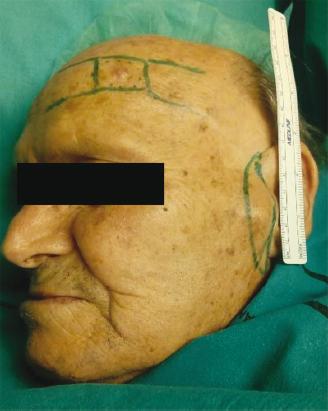
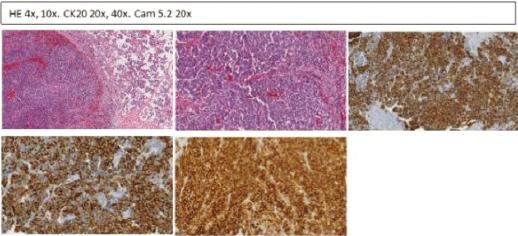
El primer caso es un varón de 87 años, asintomático, con antecedentes de tabaquismo activo, hipertensión arterial, dislipidemia, claudicación intermitente e hipertrofia benigna de próstata. Es remitido desde la consulta de dermatología por presentar, en la región frontal izquierda, una lesión rosada pápulo-tumoral de 1.7 cm de diámetro de 3 meses de evolución, con sospecha clínica de carcinoma basocelular vs. angioqueratoma (Fig. 1). Se realiza una biopsia que arroja el resultado de CCM. En la siguiente revisión se objetiva un nódulo en la región preauricular homolateral, y se realiza punción-aspiración con aguja fina que indica posible metástasis ganglionar intraparotídea. El estudio de extensión con tomografía computarizada no mostró otros hallazgos relevantes, conformando un estadio IIIB (T1N1bM0). Se realizó ampliación de márgenes y parotidectomía superficial sin incidencias, por lo que fue dado de alta. En el análisis anatomopatológico se identificó una lesión de 0.6 × 0.4 cm, con patrón de crecimiento infiltrativo, que llegaba a la dermis y la hipodermis, respetando los márgenes quirúrgicos de resección. El espesor tumoral medido desde el estrato granuloso hasta el punto de máxima infiltración fue de 4 mm. Respecto al tumour Infiltrating lymphocytes (TIL), se observaba un infiltrado linfocitario difuso peritumoral en la base. Las células presentaban un núcleo redondo, vesicular, con cromatina densa, múltiples nucleolos y un citoplasma escaso. El contaje mitótico fue de 5-6 mitosis/cga, con un índice de proliferación celular Ki67 > 90%. En cuanto al perfil inmunohistoquímico, expresaba CK20 (en gota), CAM5.2, enolasa, sinaptofisina y neurofilamentos, y era negativo para TTF1 (Fig. 2). La parotidectomía superficial mostró metástasis en un ganglio linfático intraparotídeo con extensión tumoral periganglionar (Fig. 3). A los 12 meses tras el diagnóstico no ha presentado recidivas ni progresión de la enfermedad.

Caso 2
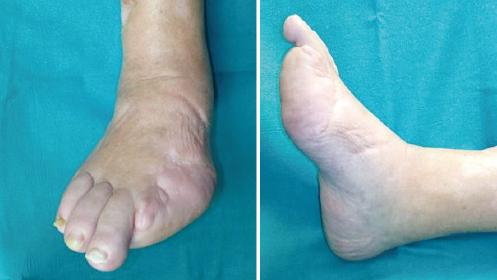
El segundo caso es una mujer de 78 años con antecedentes de hipertensión arterial, diabetes mellitus tipo 2 de mal control, insuficiencia cardíaca y cardiopatía isquémica estable. Presentaba una lesión tumoral eritematosa de superficie lisa en el dorso del primer dedo del pie derecho, de 6 meses de evolución y asintomática. Fue biopsiada con la sospecha clínica de linfoma vs. melanoma amelanótico (Fig. 4). En un segundo tiempo se realizó extirpación y biopsia selectiva de ganglio centinela (BSGC) inguinal derecho. En los hallazgos anatomopatológicos se observó una tumoración de 3.9 cm, de patrón de crecimiento infiltrativo, con invasión vascular y tumor en margen quirúrgico, compatible con CCM. El espesor tumoral era de al menos 20 mm. La reacción linfocitaria que se apreciaba era escasa y se situaba en todo el componente invasivo del tumor. El resto de las características histológicas e inmunohistoquímicas eran similares a las descritas en el caso previo. Además, se identificó metástasis ganglionar sin extensión extranodal en el ganglio centinela inguinal, conformando finalmente un estadio IIIA (pT4N1aM0). Presentado el caso en el Comité de Tumores, se decidió realizar ampliación de márgenes, que fue negativa para enfermedad residual, y vaciamiento inguinal derecho, que reveló metástasis subcapsular en uno de diez ganglios remitidos (Fig. 5). La paciente fue dada de alta del servicio de cirugía plástica y reparadora a los 3 meses al no presentar complicaciones ni enfermedad residual, para seguimiento por parte de la consulta de dermatología, sin objetivar recurrencia de la enfermedad a los 18 meses tras el diagnóstico, con un Eastern Cooperative Oncology Group (ECOG) de 4 (Fig. 6).


Se ofreció radioterapia adyuvante a ambos pacientes, pero no desearon recibirla.
Discusión
El diagnóstico clínico del CCM supone un reto por su baja frecuencia y su similitud clínica con otras lesiones, lo que supone que solo se plantee como primera opción diagnóstica en el 1% de los casos. Se ha propuesto el acrónimo AEIOU (Asintomático, Expansión rápida, Inmunosupresión, mayores de 50 años [Old] y exposición Ultravioleta)5 para valorarlo como posibilidad diagnóstica en aquellos casos que reúnan más de tres características.
En la patogénesis del CCM interactúan factores genéticos, moleculares y ambientales. Por un lado, se ha implicado la RUV, basándose en la mayor incidencia en regiones geográficas con niveles más altos de exposición y en su tendencia a aparecer en zonas fotoexpuestas (la mitad aparecen en la cabeza y el cuello)1, siendo 25 veces más frecuente en pieles blancas y a menudo hallándose adyacente a otras lesiones asociadas a la RUV, habiéndose objetivado mutaciones de citosina a timina
(indicativas de daño solar), especialmente en los tumores MCPyV negativos1,6.
Un 60 - 97% de los CCM son MCPyV positivos, quedando caracterizado por primera vez este poliomavirus ubicuo en muestras tumorales de CCM en 20087, demostrándose posteriormente su papel en la oncogénesis mediante la integración clonal en el genoma6. Asimismo, la inmunosupresión aumenta el riesgo del desarrollo de CCM, y la inmunosenescencia podría explicar el aumento de la incidencia de CCM con la edad6.
Debido a la baja incidencia de este tumor y la edad avanzada de los pacientes, no existe un algoritmo terapéutico establecido, pero se acepta que en los estadios I-III el tratamiento debe estar dirigido al tumor primario y al lecho ganglionar regional, siendo de primera elección la cirugía con márgenes amplios (1 - 3 cm)8. Debe explorarse el área ganglionar correspondiente en todos los pacientes, si es posible con ecografía. En caso de no presentar adenopatías sospechosas de malignidad, debe realizarse BSGC, ya que hasta en un tercio de estos pacientes se objetivará enfermedad ganglionar microscópica9.
La presencia de metástasis ganglionares ocurre en el 30% de los pacientes en el momento del diagnóstico10 y hasta en un 80% en el transcurso de la enfermedad, siendo el principal predictor de supervivencia, junto con el estadio clínico1. En caso de existir adenopatías clínicamente palpables o radiológicamente sospechosas, deberá realizarse estudio anatomopatológico. El tratamiento de las metástasis ganglionares es la linfadenectomía. En estos casos, está indicado un estudio de extensión.
La radioterapia adyuvante del lecho tumoral está indicada en todos los casos (en especial si hay márgenes afectos, grandes tumores, ganglios clínicamente positivos y factores de mal pronóstico histológico)9, ya que el CCM es muy radiosensible. Tras la linfadenectomía se puede plantear radioterapia del territorio de drenaje. En los casos en que sea negativo se recomienda observación debido al bajo porcentaje de falsos negativos de esta técnica8. Se acepta que la radioterapia puede disminuir la recidiva locorregional y mejorar la supervivencia global, aunque hasta la fecha no existen estudios aleatorizados que lo demuestren. La radioterapia como modalidad única de tratamiento se realiza en pacientes no candidatos a cirugía9.
Los factores de mal pronóstico incluyen los márgenes quirúrgicos afectos (el más importante de cara a la recurrencia local), la edad superior a 70 años, el sexo masculino, el tamaño superior a 2 cm, la localización en las extremidades y posiblemente los estados de inmunosupresión4,10. Por otro lado, los tumores MCPyV positivos presentan un pronóstico más favorable. Los títulos de anticuerpos frente al MCPyV se correlacionan con la carga tumoral y se modifican en respuesta al tratamiento, por lo que se han propuesto para monitorizar la recurrencia11, aunque esta determinación no está disponible en nuestro centro. No existen en la actualidad otros marcadores inmunohistoquímicos de mal pronóstico reconocidos.
En cuanto a los pacientes con enfermedad a distancia (estadio IV), se requiere un manejo multidisciplinario. En primera línea se sitúa la inmunoterapia, que incluye los inhibidores de PD-L1 (avelumab) y de PD-1 (pembrolizumab y nivolumab)10, que se ha visto en ensayos clínicos no aleatorizados que mejoran las tasas de respuesta mantenida con respecto a la quimioterapia convencional (ciclofosfamida, doxorubicina, vincristina, etopósido, cisplatino), la cual puede ser una alternativa en determinados pacientes (contraindicación de inmunoterapia, etc.). Es preferible la inclusión en un ensayo clínico cuando sea posible10. Futuras vías de abordaje terapéutico incluyen los fármacos antivirales, dada la relación con el MCPyV.
Las recidivas ocurren en los primeros 3 años en el 90% de los casos, por lo que las últimas guías10 recomiendan seguimiento cada 3-6 meses durante los primeros 3 años tras el diagnóstico y posteriormente cada 6 - 12 meses. La mejor prueba para la detección de metástasis es la tomografía por emisión de positrones; por orden de frecuencia, las metástasis afectarán la piel, los pulmones, el hígado, los huesos, el cerebro, la médula ósea y otros órganos12.
Conclusiones
– El CCM es una afección rara y agresiva que se presenta como un nódulo eritematoso de rápido crecimiento y asintomático en zonas fotoexpuestas de pacientes de edad avanzada.
– En su patogenia están implicados la RUV, la inmunosupresión y el MCPyV.
– La cirugía es el pilar fundamental del tratamiento locorregional del CCM.
– Ocho de cada diez pacientes presentarán afectación ganglionar en el transcurso de la enfermedad, siendo uno de los principales factores pronósticos.
– Si no existen adenopatías clínico-radiológicamente reconocibles, debe realizarse BSGC preferiblemente en el mismo acto quirúrgico; si existen adenopatías o la BSGC es positiva, está indicada la linfadenectomía.
– La radioterapia se acepta como adyuvancia en todos los estadios y ha demostrado un mejor control locorregional.
– En la enfermedad a distancia es de primera elección la inmunoterapia y la participación en ensayos clínicos.

