











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de hipermovilidad articular (SHA) es un desorden hereditario que se caracteriza por hiperlaxitud articular y dolores musculoesqueléticos. El término hipermovilidad se refiere al incremento en los movimientos activos o pasivos de las articulaciones con base en sus rangos normales.1
El diagnóstico se determina por el puntaje de Beighton, el cual habla de una alta especificidad de la enfermedad. Este puntaje contempla la flexibilidad en cinco áreas del cuerpo (columna/caderas, codos, quintas metacarpofalángicas, pulgares, muñecas y rodillas, obteniendo un puntaje de hasta nueve puntos, con una especificidad alta (80%).2
La importancia de este artículo es proveer información actualizada sobre el SHA, ya que suele ser un padecimiento muy frecuente con reportes de 45% en pacientes que cumplen los criterios de diferentes clínicas reumatológicas,3 principalmente en población infantil y adolescente y para el cual es común que exista un retraso en el diagnóstico y tratamiento.
Antecedentes
Este grupo de síntomas fue considerado como enfermedad hasta 1967 por Kirk y colaboradores,3 quienes contemplaron síntomas musculoesqueléticos y la presencia de hipermovilidad articular generalizada.4,5 Es importante realizar un adecuado diagnóstico diferencial para descartar alguna otra enfermedad reumática.4
En los años 80 se empezó a observar un fenotipo sobrepuesto con enfermedades del tejido conectivo, principalmente a nivel cutáneo u óseo.4 En los años 90 se estudió el dolor crónico y las disautonomías que tenían las personas con SHA, llegando a la conclusión de que son complicaciones de la enfermedad;6,7 igualmente se relacionó el aumento en problemas ginecológicos y gastrointestinales.8
Las primeras escalas que evaluaron la hipermovilidad articular fueron los criterios de Beighton, método utilizado durante 30 años;3 y el síndrome de hipermovilidad formó parte de los criterios de Brighton publicados en 1998.9
Definición
El síndrome de hipermovilidad articular es un trastorno sistémico, hereditario del tejido conectivo asociado a hipermovilidad articular generalizada, en el cual una o más articulaciones sinoviales se mueven más allá de los límites normales, con presencia de dolor articular difuso9,10,11 y sin enfermedad del tejido conectivo subyacente.9 El mecanismo bajo el cual los síntomas articulares se desarrollan no se ha entendido del todo. De acuerdo con algunos estudios, los pacientes con SHA tienen hipermovilidad articular así como dolor articular crónico y otros signos neuromusculoesqueléticos relacionados con un defecto en el colágeno.9
El SHA es conocido también como síndrome de hipermovilidad articular benigno. El término benigno se utiliza para distinguirlo de otras condiciones más severas como Ehler-Danlos (tipo clásico o vascular), síndrome de Marfan y osteogénesis imperfecta que también se presenta con hipermovilidad articular y en la que puede descartarse el diagnóstico de SHA.9
La hipermovilidad se define cuando un rango de movimiento que excede a lo que se considera normal para esa articulación, tomando en cuenta la edad del paciente, género y grupo étnico. No es un estado de enfermedad ni se considera un diagnóstico, es decir, la hipermovilidad articular se convierte en síndrome cuando se le atribuye el surgimiento de los síntomas; el dolor y la inestabilidad son los más importantes.
Existe una confusión entre el uso de los términos hipermovilidad articular y el síndrome de hipermovilidad articular.12
Epidemiología
El SHA es un rasgo hereditario común,10 se presenta frecuentemente en mujeres asiáticas en 20-40%.3 En hombres se observa en 10-30%.13
El rango de prevalencia del síndrome de hipermovilidad articular es de 4 a 38.5%. Esto ha sido tema de muchos estudios, la investigación inició a principios de la década de los 60. Está bien establecido que el síndrome de hipermovilidad articular puede predisponer al desarrollo de dolor articular en niños.9
Se estima que entre 10 y 15% de niños sanos con articulaciones hipermóviles se asocia el término de síndrome de hipermovilidad articular únicamente cuando no se encuentra otra causa de su dolor.14
En general, la prevalencia del síndrome de hiperlaxitud reportada en la literatura es variable, pero se sabe que es mayor en las mujeres y en individuos de ascendencia africana y asiática, con menor cantidad de síntomas conforme aumenta la edad.15 La prevalencia en poblaciones sanas, usando los criterios de Brighton, se ha reportado entre 15 y 17.9% en adolescentes y atletas jóvenes del sexo femenino. En un estudio realizado por Russek y Errico se encontró que la prevalencia del SHA no cambia significativamente con el nivel de actividad.16
Factores de riesgo
Los factores de riesgo incluyen la edad del paciente, etnicidad, actividad física, hábitos físicos y traumas así como la facilidad del observador para evaluar la movilidad articular con los criterios de Brighton.10
De acuerdo al estudio de Adib y colaboradores se observó en los antecedentes familiares que quienes tienen familiares de primer grado que padecen de hipermovilidad articular, ésta se presentará en 63% y en 27% de los familiares de segundo grado.17
Numerosos estudios han informado que la prevalencia de trastornos temporomandibulares es mayor en los sujetos con síntomas de hipermovilidad articular, lo cual sugiere que la hiperflexibilidad puede ser un factor de riesgo asociado positivamente con pacientes que padecen trastornos temporomandibulares.15
La hipermovilidad articular en general es más común en caucásicos versus afroamericanos. Aunque no hubo asociaciones entre hipermovilidad y factores de razas múltiples con cambios cromosómicos variados, la asociación entre la hipermovilidad y los síntomas en columna pueden diferir por la raza. En un estudio se reportó que los caucásicos con hipermovilidad eran más propensos a tener síntomas lumbares que aquéllos sin hipermovilidad.18
Otros factores que afectan la articulación y rango de movimiento incluyen cambios bioquímicos en la estructura de colágeno y elastina que causan una pérdida de resistencia a la tracción que conduce a la laxitud y al aumento de movilidad de las articulaciones.15
Etiología
El síndrome de hipermovilidad es parte de un grupo de trastornos hereditarios del tejido conectivo que incluyen el síndrome de Marfan, Ehlers-Danlos y osteogénesis imperfecta. Comparten una sobreposición fenotípica con estos trastornos que se le ha llamado síndrome de Ehlers-Danlos tipo hipermovilidad, pero históricamente se le ha considerado una forma más benigna.19
Varios estudios han demostrado que el SHA tiene un fuerte componente genético con patrón autosómico dominante; familiares de primer grado con el trastorno pueden identificarse en 50% de los casos. Mutaciones en el gen de la fibrilina han sido identificadas en familias con SHA.9
Las personas con SHA pueden experimentar síntomas referentes a respuesta reactiva anormal del sistema nervioso autónomo, particularmente aumentos de la frecuencia cardíaca en reposo, lo que puede manifestarse como un síntoma de ansiedad.20
El SHA/EDS-HT es difícil de reconocer debido a la ausencia de hallazgos físicos específicos y a que no existen marcadores genéticos específicos, a excepción de unos cuantos casos aún controvertidos con mutaciones en la tenascina XB y el gen de colágeno tipo III alfa 1. En consecuencia, el JHS/EDS-HT sigue siendo un diagnóstico de exclusión basado en criterios de diagnóstico clínico internacionalmente aceptados. Sin embargo, de acuerdo a Gharbiya y colaboradores es urgente la necesidad de revisar los criterios existentes.21
Curiosamente, algunas osteocondrodisplasias autosómicas dominantes (seudoacondroplasia y algunos casos de displasia epifisaria múltiple) son causadas por mutaciones de las glicoproteínas COMP que interfieren con el conjunto de matriz extracelular normal, que se cree contribuyen al desarrollo de los fenotipos de la enfermedad,22,23 es decir, la hipermovilidad pronunciada y los niveles bajos de COMP suero son características de estas osteocondrodisplasias.24
Las seis proteínas identificadas incluyen APOB y TTR así como las cuatro proteínas relacionadas del complemento. La concentración plasmática de APOB es conocida por ser un buen marcador de riesgo cardiovascular. APOB es un ligando para el receptor de lipoproteína de baja densidad (LDL) que participa en el transporte de colesterol a los tejidos periféricos y origina su acumulación en la pared arterial. Los niveles elevados de la apolipoproteína AI (APOA1) y APOB se han observado también en pacientes con osteoartritis.
Los cambios en los niveles de expresión de algunos factores del complemento en pacientes con SHA, en comparación con aquéllos de los individuos control, pueden contribuir a los síntomas de dolor crónico.25
Calidad de vida y discapacidad
Las cinco principales determinantes para la disminución de la calidad de vida son: deterioro físico, dolor crónico, fatiga, mala adaptación cognitiva (es decir, quinesiofobia) y angustia sicológica.10
Existe un gran número de estudios que demuestran la asociación entre el síndrome de hipermovilidad articular y quejas de salud crónicas en los adultos. Mientras que las artralgias y el dolor regional crónico son los síntomas predominantes, hay muchas manifestaciones extraarticulares con disautonomía y neuropatía periférica, hernia, prolapso uterino/rectal, depresión y ansiedad, por nombrar algunos.17
Otros autores apoyan el concepto de que la hipermovilidad tiene un fenotipo multisistémico y se sugieren posibles mecanismos que median la vulnerabilidad clínica de los síntomas neurosiquiátricos.26
En un estudio reciente se demostró un grado de discapacidad en miembros inferiores comparable con los pacientes que padecían osteoartritis, observado durante 10 años.27 Esto refleja una disfunción temprana biomecánica que afecta la marcha y postura, asociada con un patrón anormal de la activación de músculos en ejercicios estáticos y dinámicos. Además, pueden presentar quinesiofobia que se define como el miedo a moverse debido al dolor crónico, incrementando el deterioro muscular ocasionando así deterioro físico y agotamiento general.
La discapacidad es multidimensional, está influenciada también por factores sicológicos y sociales a consecuencia de la enfermedad.
El síndrome del dolor crónico es consecuencia de la ansiedad y depresión por mal manejo terapéutico.10 El síntoma primario del síndrome de hipermovilidad articular es el dolor, que inicialmente ocurre en la forma de episodios agudos y se vuelve crónico con el tiempo. Los pacientes desarrollan dolor crónico como resultado de trauma en las articulaciones, músculos y ligamentos durante las actividades diarias, en combinación con la laxitud articular excesiva. La calidad del sueño, las relaciones sociales, las actividades físicas y la calidad de vida se ven adversamente afectadas por la presencia de dolor persistente.28
Para muchos, si no es que en todos los pacientes con síntomas de SHA, en la niñez éstos son relativamente leves y discretos, otros pacientes presentan dolores más persistentes en músculos y con demasiado estrés y continuarán con dificultades hasta que su musculatura esté fortalecida por ejercicios apropiados o como resultado del crecimiento en la pubertad y adolescencia. Hay, sin embargo, un grupo severamente debilitado y deshabilitado cuyas vidas y educación son interrumpidas debido al desarrollo de dolor crónico secundario.12
Los resultados del metaanálisis de tres estudios indicaron que existe cuatro veces mayor probabilidad de ansiedad en las personas con el síndrome de hipermovilidad articular en comparación con los controles. Además, parte de los resultados del metaanálisis indicaron una diferencia estadística significativa entre los grupos, con mayor gravedad de los síntomas de ansiedad en las personas con síndrome de hipermovilidad articular en comparación con el grupo control.20
Entre los pacientes con síndrome de hipermovilidad articular hay una incidencia aumentada de síntomas siquiátricos, incluyendo depresión, ansiedad y trastornos de pánico debido a años de sufrimiento por los efectos secundarios de la condición como artralgias de larga duración, alteración en la calidad del sueño y fatiga.28
Las características de la enfermedad y las situaciones que viven los pacientes pueden propiciar dificultad para comer, esto representa una pérdida de peso e incluso trastornos de la alimentación y por ende, una mala nutrición.29
Las características clínicas de estos pacientes tienden a superponerse con las del trastorno de coordinación del desarrollo y pueden asociarse con el aprendizaje y otras discapacidades. Las consecuencias físicas y sicológicas de estas dificultades adicionales se suman a las principales manifestaciones del trastorno preexistente del tejido conectivo, afectando el bienestar y el desarrollo de los niños y sus familias.30
Manifestaciones clínicas
El inicio de los síntomas puede ocurrir a cualquier edad. Típicamente, los niños tienen dolor autolimitado en múltiples articulaciones; sin embargo, el dolor puede durar un tiempo prolongado y puede volverse constante en el adulto.9
Los tipos de dolor que suelen presentar son el de tipo neuropático (neurogénico) y nociceptivo (inflamatorio). El nociceptivo es la sensación sicológica generada por el daño tisular o la alteración en los receptores de dolor. El neuropático es el dolor relacionado directamente con lesión del sistema nervioso, involucrando la transmisión y la modulación. Además éste presenta un subtipo, el cual es disfuncional, siendo de tipo orgánico como en la enfermedad intestinal funcional, fibromialgia y cefalea ocasionado por una exacerbación anormal de la intensidad del desarrollo del dolor por estímulos viscerales y externos del sistema nervioso central.10 El dolor puede involucrar cualquier articulación, pero es más común la rodilla y el tobillo, la actividad física repetitiva igualmente exacerba el dolor.9 La fatiga y el dolor generalizado son asuntos comunes con hiperestesia resultante de una nociocepción aumentada de los tejidos estirados como una explicación probable; el dolor con frecuencia resulta en inactividad con descondicionamiento que exacerba los síntomas.13 Especulativamente, posibles mecanismos incluyen la susceptibilidad de los individuos con hipermovilidad elevada a (amenaza de) dolor y/o una perturbación de control autonómico. Las diferencias en la actividad de la amígdala se producen en trastornos de dolor, incluyendo la fibromialgia, síndrome del intestino irritable y síndrome de dolor regional crónico.26
La hipermovilidad articular representa un factor de riesgo de dolor musculoesquelético durante la adolescencia, que comprende una distribución específica, es decir, el hombro, la rodilla y el tobillo/pie. Estas relaciones fueron más fuertes en presencia de obesidad, lo cual es consistente con una vía causal por la cual la hipermovilidad articular conduce a dolor en sitios expuestos a mayores fuerzas mecánicas.31 Síntomas menos frecuentes son rigidez, mialgias, calambres y dolor no articular en miembros.9
Las articulaciones que con más frecuencia se ven afectadas son el cuello y los hombros, normalmente de presentación simultánea. La inestabilidad del hombro se puede ver involucrada cuando el cuello está afectado, por lo que tiene limitación a la movilidad por el riesgo de subluxación o luxación, evitándose así la realización de actividades.23 La cervicalgia puede ocurrir como resultado de enfermedad en el disco cervical, puntos miofasciales desencadenantes o hipermovilidad de la columna cervical.32 La hipermovilidad cervical y la disfunción articular temporomandibular son factores que predisponen el desarrollo de la cefalea. La malformación de Chiari tipo I, es decir, inestabilidad de la articulación occipitoatlantoaxial, se incluye en el SHA.10 De acuerdo a estudios de doble corte la prevalencia de hiperlaxitud, está asociada a 18% con casos de mano (en especial articulación interfalángica proximal), osteoartritis de la rodilla y disminución de los niveles de suero oligomérico de matriz del cartílago con un predominio en sexo femenino. Estos resultados se validaron adicionalmente con estudios radiológicos en sus diferentes poblaciones.24
La escoliosis es un hallazgo que con frecuencia se observa en niños y adultos con SHA. Un estudio reciente de laxitud articular durante el screening de escoliosis de 1,273 niños (598 hombres; 675 mujeres; promedio de edad 10.4 años) con el uso del escoliómetro de Bunnell mostró una correlación entre la escala de Beighton y la rotación del tronco de > 7o.12
Los reportes de investigación describen alta prevalencia de síntomas como alteraciones del sueño, fatiga, intestino irritable, torpeza, ansiedad y depresión entre los pacientes con SHA.16 El SHA comparte muchos síntomas con los trastornos funcionales gastrointestinales, incluyendo trastornos de sueño, fibromialgia, trastornos sicológicos, cefalea migrañosa así como un perfil demográfico similar.19 También se asocia con enfermedad intestinal funcional y fibromialgia.10
Existe interés particular en la asociación entre enfermedad celíaca, trastornos del tejido conectivo y artritis reumatoide, los dos últimos trastornos con trasfondo genético. El involucro al tejido conectivo puede tener implicaciones en trastornos gastrointestinales, estructurales y funcionales. Diferentes investigaciones han mostrado la presencia de síntomas como dolor abdominal, hinchazón, náusea, reflujo, disfagia, vómito y constipación. La enfermedad de Ehlers-Danlos y el síndrome de hipermovilidad articular se han asociado a trastornos gastrointestinales incluyendo dispepsia, reflujo gastroesofágico, síndrome de intestino irritable, constipación funcional, constipación por tránsito intestinal lento, incontinencia fecal, retardo en vaciamiento gástrico y enfermedad de Crohn. En un estudio realizado en 2016 por Laszkowska y colaboradores se encontró asociación entre enfermedad celíaca y Ehrles Danlos y síndrome de hipermovilidad articular.33 El dolor abdominal inexplicable se manifiesta en 86% de los pacientes con síntomas como reflujo, náuseas, vómito, diarrea, constipación, distensión y pirosis. Se desconoce la fisiopatología por la cual se presenta tan severa afectación gastrointestinal.10
Pacientes que muestran síntomas inexplicables gastrointestinales y dismotilidad intestinal normalmente se clasifican como alteraciones funcionales gastrointestinales que incluyen dispepsia y constipación funcional.8 A pesar de que los síntomas gastrointestinales superiores dependían de factores de dolor autonómicos y crónicos, muchos de los síntomas se incrementan al aumentar la gravedad del fenotipo JHS.34 La asociación de SHA o prolapso de válvula mitral con fibromialgia ha sido estudiada muchas veces en la literatura. Un estudio realizado por Kozanoglu y su equipo en 75 mujeres con fibromialgia reveló que el síndrome de hipermovilidad articular y el prolapso de válvula mitral fueron comunes en pacientes con fibromialgia y el síndrome de hipermovilidad articular sirve como factor de riesgo de prolapso de válvula mitral entre pacientes con fibromialgia.35
La disfunción del sistema nervioso autónomo también se ha reportado en pacientes con SHA que han sido sometidos a estudio.19 Las manifestaciones gastrointestinales como constipación crónica, reflujo gastroesofágico, dolor abdominal crónico y síndrome de colon irritable son comúnmente reportados en SHA, al igual que los síntomas de síncope, mareo e hipotensión ortostática. Estas características juntas con frecuencia se citan como evidencia de disfunción autonómica en SHA.13
Las anomalías oculares también se han incluido como un signo menor de acuerdo a los criterios de diagnóstico para JHS; sin embargo, se han publicado muy pocos artículos sobre este tema. Se incluye el síndrome de Stickler, que se caracteriza principalmente por miopía severa, degeneración vitreorretiniana y cataratas. Mishra y colegas reportaron por primera vez una alta incidencia en laxitud del párpado y la inclinación palpebral antimongoloide evaluando a 34 pacientes. Otras alteraciones que también pueden presentarse son la ptosis unilateral, disco óptico inclinado y la xeroftalmia por deficiencia de la película lagrimal.21
Los trastornos en el sueño ocurren en 37% de los niños con SHA, mientras que movimientos periódicos de extremidades se observan en 67% de los adultos con enfermedad de Ehlers-Danlos.32
Varios estudios sugieren que la migraña es más frecuente en pacientes con SHA. Hakim y colaboradores encontraron que la prevalencia de migraña es de 40% en pacientes con SHA y de 20% en la población general. En un estudio realizado por Bendik y su equipo con una muestra de 28 casos y 232 controles se detectó una prevalencia de migraña de 75% en pacientes con SHA y de 43% en el grupo control. Migraña sin aura y migraña típica con aura fueron diagnosticadas en 68 y 32% de los pacientes en el grupo de SHA y de 39 y 10% del grupo control, respectivamente. El cambio en el tejido conectivo en SHA puede ocasionar reactividad vascular que predispone al desarrollo de migraña, también es posible que esté asociado a disautonomía que ocasiona migraña como el síndrome de taquicardia postural ortostática; exámenes autonómicos revelan evidencia de hiperactividad a y b adrenérgica en pacientes con SHA.32
SHA en niños
El SHA es el trastorno reumatológico que por excelencia trasciende la división niño-adulto. En el momento del nacimiento una de las más tempranas y conocidas asociaciones con hipermovilidad llamada displasia congénita de cadera, manifestada por una cadera «que truena» o como una dislocación congénita franca que puede ser aparente, debe siempre considerarse con examen clínico y, cuando sea apropiado, analizarse por ultrasonido. Los criterios de Brighton, hablando estrictamente, no han sido validados por completo en niños.12
Una de las características más frecuentes del SHA en edades tempranas es el retraso motor. Se observó que aproximadamente en un tercio de los niños con hipermovilidad articular generalizada en una de las series a veces hay torpeza del movimiento (que persiste en la edad adulta) así como inquietud irrepresible. Cuando empiezan a caminar, los niños pequeños hipermóviles caminan de puntas o con marcha con los dedos hacia adentro o hacia fuera, lo que feruliza los tobillos. Esto les da la sensación de mejor soporte y así emplean una marcha anormal. Sin embargo, la marcha en puntas frecuentemente ocasiona caídas cuando los pies tropiezan sobre sí mismos.4
Una vez que caminan, los padres notan que sus hijos tienen pies planos y pronados, lo cual conlleva a marcha anormal con características como patelas «bizcas», torsión tibial, anteversión femoral e hiperlordosis.
Se ha investigado a muchos niños hipermóviles con equimosis, que es una característica común en esta condición. Los exámenes de tiempo de sangrado son por lo regular normales y las equimosis pueden ser explicadas por una falta de soporte capilar normal permitida por un colágeno inherentemente débil.
Criterios de signos mayores y menores
De acuerdo al estudio de Remving y colaboradores se mencionan los criterios que presenta el SHA, los signos principales incluyen signos cutáneos (71%), luxación (es/subluxación (s) (64%) y artralgia (57%). Los signos secundarios menores son oculares (79%), gastrointestinales (79%), reumatismo de tejidos blandos (79%), signos de disautonomía (64%), venas varicosas, etcétera (57%) y signos dentales (57%). La presencia de un mínimo de dos signos principales era obligatoria de acuerdo con ocho panelistas (57%), tres signos principales según tres panelistas (21%) y cuatro signos principales según un panelista. Se requiere un mínimo de dos signos principales que son suficientes para cumplir con los ciriterios establecidos de EDS-HT.36
Diagnóstico
Para diagnosticar el síndrome de hipermovilidad por lo general se aceptan los criterios de Brighton, los cuales fueron publicados en 1998.9 No hay estudios de imagen, laboratorio o genéticos para SHA. A pesar de que los criterios de Brighton han sido criticados por no ser lo suficientemente consistentes o confiables, son los únicos que en la actualidad están aceptados a nivel internacional como criterios diagnósticos de SHA (Figura 1).16
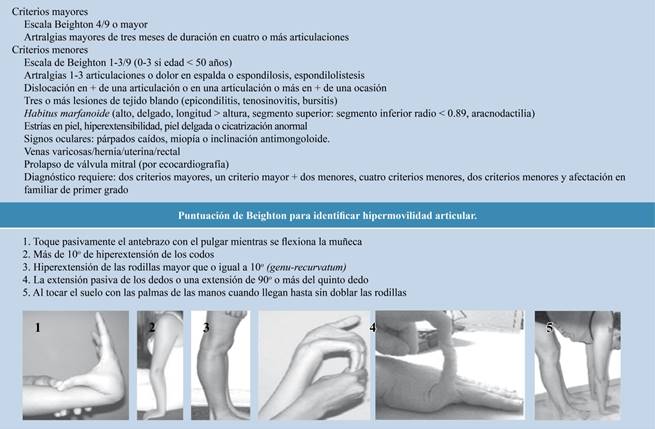
Tratamiento
Dos ensayos randomizados controlados han demostrado una reducción de 30-40% del dolor en niños con SHA después de seis a ocho semanas de un programa de ejercicios supervisados por fisioterapista.13 La fisioterapia se considera la estrategia más eficaz, incluye ejercicios de fortalecimiento, mejora de propiocepción y estiramientos suaves. Sin embargo, tiene sus límites de acuerdo al tiempo empleado, variabilidad entre operadores y eficiencia a largo plazo; las alternativas para ser más eficaz son terapias sicológicas, principalmente la terapia cognitivo conductual (TCC) masajes y tratamientos no convencionales.
Para el manejo del dolor crónico se puede coadyuvar con medicamentos como amitriptilina, duloxetina y SSRI/ SNR. Un porcentaje de 10% de proloterapia con dextrosa demostró ser eficaz para reducir el dolor a la palpación en pacientes con complicaciones de SHA temporomandibular.9
La cirugía no es una opción para mitigar el dolor. Tratamientos pasivos como hielo, masaje, electroterapia, reforzamiento y ferulización pueden ser útiles en los momentos de exacerbaciones de dolor, pero deben evitarse como estrategias a largo plazo. Analgésicos como paracetamol pueden ser beneficiosos en crisis de exacerbación de dolor; sin embargo, los antiinflamatorios no esteroideos y los opiáceos con frecuencia son mal tolerados en esta población debido a la exacerbación de los síntomas gastrointestinales o la fatiga, respectivamente. Hay cierta evidencia de que el uso a largo plazo de opiáceos (> 16 semanas) puede empeorar el dolor crónico y causar dependencia.
Algunos pacientes con dolor crónico pueden beneficiarse de terapias farmacológicas más allá de la analgesia para modificar la percepción del dolor como inhibidores selectivos de la recaptura de serotonina, antidepresivos tricíclicos y antiepilépticos como gabapentina, pero estas drogas deben utilizare con cautela y no en un término a largo plazo.13
Diagnósticos diferenciales
El síndrome de Ehlers-Danlos tipo hiperlaxitud se define por hipermovilidad articular generalizada (puntuación de Beighton 5/9) y por una sensación que describen los pacientes como suave, «aterciopelada»’ de la piel que puede o no ser apreciable como hiperelasticidad. A menudo, el síndrome de Ehlers-Danlos tipo hiperlaxitud es el «diagnóstico» «por defecto» de los niños con hipermovilidad articular, junto con el dolor generalizado y sin signos o síntomas de otros trastornos hereditarios del tejido conectivo, lo que dificulta el diagnóstico y el verdadero manejo del síndrome de hipermovilidad articular.37 El síndrome de Marfan es una enfermedad autosómica dominante, multisistémica de tejido conectivo y es una condición relativamente común, afecta a una de 5,000 personas. Es causado por defectos en el gen FBN1, este gen codifica la proteína de fibrilina e incluye mutaciones de TGFBR1 y TGFBR2. Las características principales son la afección al sistema musculoesquelético, ocular y cardiovascular. Existe laxitud de las articulaciones, dislocación, dolor articular crónico y artritis degenerativa. Su diagnóstico se basa a partir de los criterios de Ghent y genetistas.14 Expertos aciertan que SHA y el síndrome Ehlers-Danlos del tipo hipermovilidad son indistinguibles con el uso de herramientas diagnósticas actuales.16
Castori y colaboradores mencionan que aún no se puede distinguir si JHS y EDS-HT son dos trastornos distintos con un defecto molecular subyacente claramente diferente o JHS y EDS-HT son trastornos relativamente superpuestos que comparten de manera parcial su base molecular (también dentro de un patrón de herencia no mendeliano), es decir, son dos condiciones completamente superpuestas con el mismo apuntalamiento genético.38
La hipermovilidad articular se caracteriza por la laxitud de los ligamentos, puede representar un hallazgo aislado; sin embargo, ocurre en individuos con trastorno genético de proteínas de la matriz del tejido conectivo así como osteogénesis imperfecta o síndrome de Marfan. Cuando se agregan características clínicas como dolor articular y las causas genéticas ausentes, se puede referir al término hipermovilidad articular benigna. Se valora a partir de la escala de Beighton, siendo más de 4 positivo para hipermovilidad articular.31 El síndrome articular de hipermovilidad benigno es un término que aún no se ha comprendido por completo; sin embargo, se conoce que puede ser un trastorno de patrón autosómico con deficiencia de tenascina X, sin anormalidad en colágeno o relacionado a proteínas.14
La hipermovilidad articular generalizada puede encontrarse en displasias esqueléticas que afectan el tejido conjuntivo especializado, alteraciones neuromusculares debido al aumento de miopatías hereditarias y características de distrofias musculares.38
Discusión
El síndrome de Ehlers-Danlos (EDS) es una enfermedad rara que se caracteriza por exceso de flexibilidad y fragilidad de tejidos conectivos blandos, los cuales producen cambios con presencia de modificaciones en la piel, ligamentos, articulaciones, vasos sanguíneos y órganos. Los cambios en la piel incluyen: hiperextensibilidad, cicatrices papiráceas-atróficas en sitios de daño repetitivo, seudotumores moluscoides en la superficie extensora de codos y rodillas y nódulos subcutáneos más apreciables en extremidades, que son la tétrada cutánea patognomónica del SED clásica de acuerdo a la nosología de Villefranche. Se desarrolla a partir de la mutación COL5A1o COL5A2. Se diagnostica a partir de criterios Brighton y Villefranche.38,39 Se divide en seis tipos; sin embargo, los más comunes son hipermovilidad benigna y la clásica.22
La identificación de JHS en estos pacientes a través de la evaluación clínica simple puede ayudar a caracterizar a estos pacientes y proporcionar un diagnóstico unificador para todos sus múltiples síntomas, evitando así numerosas derivaciones innecesarias a varios especialistas. En un estudio reciente, 50% de los pacientes esperaron más de 10 años un diagnóstico de JHS.34 Algunos estudios proponen que la evaluación de rutina para la hipermovilidad articular debe incluirse en la evaluación de todos los pacientes que asisten a una clínica de reumatología, ya que la gran mayoría de los pacientes muestran una suma de sintomatologías que durante el interrogatorio orienta a otra patología; sin embargo, muchos de esos casos presentan hipermovilidad articular aunada a enfermedades reumatológicas y algunas de ellas tienen un manejo de largo tiempo por la poca capacidad de atacar el verdadero origen de muchas patologías.40
En 2005 Adib y su equipo observaron aumentada la extensibilidad en piel, menores medidas cuantitativas por ultrasonido en huesos y un incremento significativo de la excreción de productos de degradación de la colágena en orina en comparación con pacientes controles.1 Farmer y colegas mencionan que anteriormente los que presentaban SHA tenían mayor espesor de la piel; sin embargo, en su estudio encontraron que quienes eran hipermóviles tenían un espesor similar a aquéllos con movilidad normal. Además, observaron que el incremento en el estiramiento de la piel se correlaciona con la escala de Beighton, lo que sugiere que los individuos con hipermovilidad tienen mayor extensibilidad de la piel. Este diagnóstico es auxiliar y es positivo cuando el valor es ≥ 17.0%/mm.41
Existe cada vez mayor evidencia de que gran parte del dolor experimentado por pacientes, en particular niños con SHA, está asociado con el aumento de la fatiga muscular debido al desacondicionamiento y al incremento de la demanda de los músculos que intentan controlar las articulaciones con rangos hipermóviles.12
La fatiga es un síntoma deshabilitante para la mayoría de los niños con SHA. Los niños con SHA con frecuencia no reportan fatiga, a menos que se les cuestione o evalúe de manera objetiva, posiblemente por la dificultad de describirlo o porque han vivido con eso durante algún tiempo y no saben lo que es normal. La falta de sueño, la debilidad muscular y la disautonomía han mostrado asociación con empeoramiento de la fatiga en SHA.32
Se reportan complicaciones a largo plazo como inestabilidad articular, dolor musculoesquelético y cambios menores en la piel.42 Los trastornos temporomandibulares y las anomalías del olfato y del gusto son comunes entre los afectados y tienen implicaciones significativas en la alimentación.
Conclusión
El síndrome de hipermovilidad articular es un trastorno hereditario que se debe diferenciar de otras colagenopatías; muchas veces el diagnóstico resulta difícil. Es necesario distinguir las diferencias y conocer las asociaciones de las múltiples manifestaciones sistémicas involucradas en el trastorno para mejorar lo más posible la calidad de vida de los pacientes afectados.

