











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Dentro de los desórdenes congénitos de la glicosilación la osteocondromatosis múltiple (OM, OMIM 133700, 133701), también conocida como EXT1/EXT2-CDG, pertenece al grupo de las deficiencias de biosíntesis de glicosaminoglicanos asociadas a los trastornos del esqueleto y del tejido conectivo. Este grupo de entidades es infrecuente y presenta un patrón de herencia autosómico dominante, entre 70-90% de los afectados cursa mutaciones heterocigotas en los genes EXT1 localizados en 8q24.11-q24.13 o EXT2 en 11p12-p11, que codifican la exostosina glucosiltransferasas 1 y 2 (OMIM 608177 y 608210), respectivamente.1,2,3,4,5 Ambos genes son supresores de tumores, se expresan de forma ubicua, pertenecen a la familia de genes EXT y todos los miembros codifican para glicosiltransferasas encargadas en la biosíntesis del heparán sulfato,2,3 el cual interviene en el mecanismo de señalización en la placa de crecimiento del hueso endocondral y cuyas alteraciones conducirían a la formación de osteocondroma.3
La osteocondromatosis múltiple (OM) presenta una incidencia estimada de uno en 50,000 nacimientos en Europa y afecta a ambos sexos por igual.1,3,4,6 Se caracteriza por la formación de osteocondromas (tumores óseos benignos más comunes) que se encuentran cubiertos de cartílago localizados en las placas de crecimiento de los huesos largos, por lo general son indoloros y en los casos graves pueden mostrar una amplia distribución.1,3,4 Dependiendo de su número, tamaño y ubicación pueden causar complicaciones como talla baja en aproximadamente 40%, deformidad esquelética con limitación funcional, compresión de estructuras adyacentes que pueden producir dolor crónico, la aparición temprana de osteoartritis e incluso la transformación maligna a condrosarcoma entre 2 y 5% de los casos, cuyo riesgo aumenta con la edad y pueden resistir al tratamiento de quimioterapia y radioterapia.1,3,4,5,6,7,8
Se presentan dos casos de una familia con evaluación interdisciplinaria y diagnóstico de OM, en quienes se encontró una mutación sin sentido en el gen EXT1.
Casos clínicos
Caso 1
Masculino de 22 años, evaluado por presentar prominencias óseas en extremidades desde la infancia. Producto de unión no consanguínea, madre de 19 años en el momento de la concepción, la cual muestra fenotipo similar al propósito, al igual que todos los tíos maternos y abuelo materno, quien es el primer afectado en la familia (Figura 1).

Figura 1: La flecha señala el caso 1 (III:24), su madre, el caso 2 (II:13) y abuelo paterno (I:1), primer afectado en la familia. Ante una entidad con patrón de herencia autosómico dominante puede observarse que todas las generaciones y ambos sexos por igual se encuentran afectados, además se presenta trasmisión de OM de varón a varón.
Nació por vía vaginal por fórceps a las 34 semanas, de madre primigesta. Embarazo mal controlado, niega complicaciones. El peso y la talla al nacer fueron 2.500 g (DE -1.9) y 46 cm (DE -3.3), respectivamente. Evidenció adecuado desarrollo sicomotor y desenvolvimiento escolar. A los 19 años se le realizó exéresis de osteocondroma en región distal de fémur izquierdo a nivel de metáfisis, cuyo estudio histológico no presentó signos de malignidad.
Examen físico: talla 155 cm (DE -3.5); peso 42 kg (DE -3.4). Normocefalia, sin dismorfia facial de importancia. El tórax y abdomen dentro de la normalidad. En extremidades se evidencia deformidad de Madelung en antebrazo derecho, la cual produce un acortamiento mesomélico y aumento de volumen en el tercio externo de ambos antebrazos que ocasionan leve deformidad, pero no son dolorosos. Presenta alteración en la marcha.
Los estudios de radiografía simple de huesos largos de cuatro extremidades evidenciaron distorsión de la arquitectura con lesiones encondromatosas a predominio de la región metafisaria de fémur distal y tibia proximal.
Caso 2
Femenino de 42 años, (madre del caso 1) II:13 (Figura 1), evaluada igualmente desde la infancia por presentar prominencias óseas en extremidades. Producto de unión no consanguínea, de madre decimogesta, como ya se hizo referencia, su padre es el primer afectado en la familia, muestra fenotipo similar al igual que todos sus hermanos y sus dos hijos.
Menarquia a los 12 años, tres gestas, una cesárea y un aborto. A la edad de 37 años se le realizó hemipelvectomía derecha ante diagnóstico de condrosarcoma (Figura 2). Evaluación pendiente de lesión en región distal de fémur derecho.
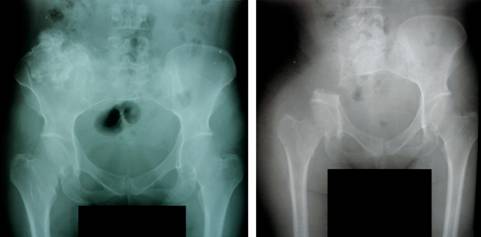
Figura 2: A la izquierda se evidencia condrosarcoma en hemipelvis derecha y a la derecha hemipelvectomía derecha.
Examen físico: talla 146 cm (DE -3.5); peso 41 kg (DE -2.1). Normocefalia, sin dismorfia facial de importancia, cuello corto, tórax y abdomen dentro de la normalidad. En extremidades se evidencia aumento de volumen en el tercio externo de ambos antebrazos que producen leve deformidad, dolor de leve intensidad y en ocasiones ausencia de dolor. Presenta igualmente alteración en la marcha.
Ambos individuos se mantienen en control por los servicios de ortopedia y traumatología, medicina interna y genética médica para asesoramiento genético familiar.
Estudio molecular
Mediante la técnica de amplificación por reacción en cadena de la polimerasa, seguida de secuenciación automática, se identificó la mutación sin sentido en heterocigosis c.1219C>T, (p.Gln407Stop) en el exón 4 del gen EXT1 en ambos casos expuestos en este artículo y en el abuelo materno.
La mutación detectada es de tipo transición por cambio de una citosina por timina en posición 1219, en una de las dos copias del gen EXT1 cambia el codón para el aminoácido glutamina en posición 407 por un codón de parada de la traducción. De traducirse, este gen mutado daría lugar a una proteína con sólo los primeros 406 aminoácidos, en lugar de 746. Por lo tanto, dicha mutación puede considerarse patogénica. Esta mutación ya ha sido descrita por Jennes y cols. y consta en las bases de datos: Multiple Osteochondroma Mutation Database (http://medgen.ua.ac.be/LOVDv.2.0/home.php) y HGMD@Proffesional 2017.4. En la segunda copia del gen EXT1 no se ha encontrado ninguna mutación, ni en ninguno de los alelos del gen EXT2.
Discusión
La OM se caracteriza por múltiples crecimientos óseos encapsulados de cartílago que surgen de la placa de crecimiento de los huesos largos o de la superficie de huesos planos. Estos osteocondromas son causados por un aumento en la proliferación de condrocitos que produce un crecimiento óseo excesivo en las metáfisis.7 Se desarrollan gradualmente durante los primeros años y crecen en tamaño y número hasta el cierre de las placas de crecimiento cuando se alcanza la maduración esquelética al final de la pubertad.4,6,7
La OM se caracteriza por una significativa variabilidad fenotípica intrafamiliar e interfamiliar7 como se evidenció en la familia expuesta en este caso, con diferentes hallazgos clínicos en los individuos estudiados. Presenta una penetrancia cerca de 100%7 y aproximadamente 10% de los casos son el resultado de nuevas mutaciones.6,7 Como ya se hizo referencia, la mutación descrita en este estudio se encuentra en las bases de datos citadas, documentada por Jennes y cols. en dos familias procedentes de Bélgica e India. El caso que nos ocupa se trata igualmente de un caso familiar con al menos cuatro generaciones de afectados procedentes de Sudamérica (Figura 1).
Los tratamientos consisten principalmente en la resección quirúrgica de las lesiones más sintomáticas y la corrección de defectos esqueléticos.4,5 Cuando los osteocondromas son difíciles de localizar, pueden presentar dolor crónico y pérdida de función que afectan la calidad de vida y las actividades cotidianas.4,6 Estudios recientes hacen mención de alteraciones extraesqueléticas en las que se incluyen dificultades sociales, en el aprendizaje, trastornos del sueño y neuropatías. La OM también puede cursar con alteraciones en el aclaramiento lipídico postprandial y en la reserva de células beta pancreáticas debido a un menor volumen,4 razón por la que se recomienda una evaluación médica interdisciplinaria incluso a edades tempranas.
En aproximadamente 50% de los individuos con OM las lesiones se diagnostican antes de los 3.5 años de edad. En casos menos severos pueden hacerse evidentes al final de la primera década. Es probable que estos casos subestimen la frecuencia de la entidad.8 En casos familiares como el expuesto en este artículo, la evaluación temprana en los hijos de algún afectado puede iniciar el diagnóstico y confirmarse con estudios de imagen.
La deformidad de Madelung presente en el caso 1 no es un hallazgo frecuente en la OM. Puede producir deformidad como arqueamiento de los antebrazos, restricción de la pronación y supinación, además de coxa valga, tibia valga y acortamiento del peroné en relación con la tibia, entre otros, por lo que ante estos hallazgos debe realizarse diagnóstico diferencial con la discondrosteosis, la forma más común de talla baja mesomélica que puede estar asociada a genu valgum y talla baja.9 Además de la ya comentada OM, en la enfermedad de Ollier, displasia epifisaria múltiple y síndrome de Turner puede evidenciarse la deformidad de Madelung,10 por tal motivo deben considerarse diagnósticos diferenciales ante la presencia de dicha alteración en el antebrazo.
Por otra parte, el riesgo de transformación maligna, aunque no es frecuente, se presentó en el caso 2. Un estudio realizado en 529 pacientes con OM no encontró relación significativa de esta complicación con la mutación en los genes EXT, sexo, gravedad de la enfermedad o número de sitios esqueléticos con exostosis. El condrosarcoma ocurrió con más frecuencia en la pelvis, la escápula y la parte proximal del fémur. Como ya se hizo referencia, ningún genotipo particular está correlacionado con la presencia de lesión maligna; sin embargo, ésta se produjo en dos pacientes relacionados con una mutación sin sentido en el gen EXT1 y fue más frecuente en individuos con historia familiar de OM,11 dos características presentes en la familia estudiada en este artículo que deben seguir siendo evaluadas en investigaciones futuras.
Por su parte, entre los diagnósticos diferenciales de OM se encuentra la ya comentada enfermedad de Ollier, entidad no familiar, con una incidencia estimada de uno en 100,000, sin predominio de sexo, caracterizada por múltiples encondromatosis y áreas de cartílago displásico, con marcada predominancia unilateral.12 Además, el síndrome de Maffucci (o enfermedad de Ollier tipo 2)13 es más infrecuente y se caracteriza por la asociación de encondromatosis y hemangiomas que puede afectar piel, mucosas y órganos internos.12 El riesgo de transformación maligna sarcomatosa puede alcanzar 25-50% en la enfermedad de Ollier y 100% en el síndrome de Mafucci.13
Se presentan dos casos familiares de OM con diagnóstico clínico por imagen y confirmación molecular, resaltando la variación intrafamiliar de la entidad, por tal motivo cada caso debe ser evaluado individual e interdisciplinariamente. De igual manera, se hace referencia a las complicaciones que pueden evidenciarse, incluso extraesqueléticas (pleitropismo) y diagnósticos diferenciales. Ante una entidad con patrón de herencia autosómica dominante, el riesgo de recurrencia es de 50% en los descendientes de un afectado y ante la presencia de un cuadro familiar es necesario hacer estudios a sus integrantes.

