











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
El género Quercus L., es el grupo más grande de la familia Fagaceae, presenta la mayor distribución y diversidad a nivel mundial, con unas 500 especies aceptadas (Valencia et al. 2016). México es considerado el centro de diversificación más grande a nivel mundial, estas especies se distribuyen en regiones montañosas del centro y noreste del país principalmente (Ramamoorthy et al. 1993, Govaerts y Frodin 1998). En México se han identificado 161 especies del género Quercus que pertenecen al subgénero Quercus, con tres secciones: Quercus, Lobatae y Protobalanus (Valencia 2004). Existe gran interés en torno a este grupo taxonómico, debido a su alta diversidad y su importancia ecológica y económica (Romero-Rangel 2006, Gual-Díaz 2014, Pérez-Mojica 2017). Debido a la variación taxonómica y morfológica dentro de la misma especie, al largo de tiempo de generación y la hibridación frecuente entre especies, se dificulta conocer el número real de especies (Villarreal et al. 2008, Sabás et al. 2017). Por lo anterior, es necesario proporcionar una solución a los problemas taxonómicos, que permita una mejor toma de decisiones para el manejo y conservación de estas especies (Valencia 2004, Hoban et al. 2009, Hipp et al. 2013). Por tal motivo, los códigos de barras de ADN proporcionan datos que ayudan a discernir relaciones taxonómicas y permiten una mejor distinción en es pecies con alto grado de similitud a niveles de detalles altamente específicos (Borek y Summer 2009, Govindaraghavan et al. 2012).
En el 2009 el Consorcio del Código de Barras de ADN (CBOL) recomendó en plantas el uso de fragmentos provenientes del genoma del cloroplasto, tales como rbcL (ribulosa-1-5 bifosfato carboxilasa/oxigenasa) y matK (maturasa K), los cuales son relativamente fáciles de amplificar debido a su estabilidad relativa, son de copia única, estructura altamente conservada y herencia uniparental (China Plant BOL Group et al. 2011). En 2011 se sugirió el uso de la región nuclear ITS2 (espaciador interno transcrito) como complemento, esta región pertenece a los genes que codifican para el ARN ribosómico (CBOL Plant Working Group et al. 2009).
La información que proporcionan los códigos de barra de ADN es utilizada con diferentes propósitos, entre ellos: hacer inventarios de biodiversidad, reconstrucción del clima y la vegetación del pasado, además, sirve como complemento de la filogenética, ya que con la integración de datos moleculares y homologías morfológicas se logra la obtención de información sobre la historia evolutiva de las especies (Hajibabaei et al. 2007, Kress et al. 2009, Costion et al. 2011). Al respecto, Hipp et al. (2013) afirmaron que es necesario establecer un método preciso, específico y universal que proporcione datos confiables para la identificación de especies y provea información sobre patrones filogenéticos en especies de Quercus. En México, actualmente sólo el 5% del total de las especies de Quercus distribuidas en el país cuentan al menos con uno de los tres códigos, lo cual representa una limitante para hacer análisis a mayor escala, y muestra la necesidad de realizar más estudios con la tecnología de código de barras de ADN con especies de distribución nacional. Por lo anterior, el objetivo de este trabajo fue determinar las relaciones filogenéticas en especies de encinos de tres regiones específicas de México, Estados Unidos y Eurasia, para demostrar el poder de identificación de tres regiones del código de barras de ADN denominadas rbcL, matK e ITS2.
Materiales y métodos
Colecta de muestras en campo
De acuerdo a datos disponibles de georreferenciación (Villarreal et al. 2008) se establecieron puntos de colecta en la región sureste del estado de Coahuila, México. Se muestrearon un total de 29 árboles. Se colectaron 5 g de tejido foliar de hojas aparentemente sanas y de brotes jóvenes los cuales se preservaron en alcohol de 96o para su análisis posterior. Los comprobantes de herbario se prepararon e identificaron morfológicamente siguiendo la guía propuesta por Villarreal et al. (2008) y se depositaron en el Herbario Antonio Narro, Saltillo, México (ANSM). Con base en los resultados de la identificación morfológica, se seleccionaron tres ejemplares por especie para el análisis genético.
Extracción, amplificación y secuenciación de ADN
Se realizaron extracciones de ADN genómico mediante el método modificado de CTAB (Sharma et al. 2003). La concentración y calidad del ADN extraído se comprobó mediante electroforesis en gel de agarosa al 0.8%, a 75 voltios durante 1:45 h, utilizando un marcador de peso molecular (1 Kb, Axygen, USA). La visualización del gel se realizó mediante el Sistema de Documentación Gel Axygen (Axygen GD-1000, USA). El ADN se conservó a -20 oC para su análisis posterior. Con el ADN genómico se llevó a cabo la amplificación de los códigos de barras usando los respectivos cebadores (Tabla 1). La reacción en cadena de la polimerasa (PCR) se realizó utilizando la mezcla comercial GoTaq Green Master Mix, 2XTM (Promega, USA), según las especificaciones del fabricante. Las condiciones de la PCR se detallan en la Tabla 1. Los productos de PCR amplificados se verificaron por medio de electroforesis en gel de agarosa (0.8%). Los productos de PCR que presentaron bandas inespecíficas, se purificaron utilizando el sistema comercial Wizard SV Gel and PCR Clean Up System™ (Promega, USA), siguiendo las instrucciones del fabricante. Se enviaron 300 ng de producto de PCR de cada muestra al servicio de secuenciación PlateSeq Service (Eurofins™, USA).
Tabla 1: Cebadores y programas de PCR utilizados para la amplificación de ADN.
| Fragmento | Cebadores (5’-3’) | Programa de PCR | Referencia |
|---|---|---|---|
| rbcL | rbcLa-F: ATGTCACCACAAACAGAGACTAAAGC rbcLa-R: GTAAAATCAAGTCCACCRCG | 94°C, 5 min; 35 x (94°C, 30 s; 50°C, 40 s; 72°C, 40 s); 72°C, 10 min; ∞4°C. | (Kress et al. 2009) |
| matK | matK-472f: CCCRTYCATCTGGAAATCTTGGTTC matK-1248r: GCTRTRATAATGAGAAAGATTTCTGC | 94°C, 5 min; 35 x (94°C, 15 s; 56°C, 20 s; 72°C, 50 s); 72°C, 10 min; ∞4°C. | (Yu et al. 2011) |
| ITS2 | ITS-2F: ATGCGATACTTGGTGTGAAT ITS-3R: GACGCTTCTCCAGACTACAAT | 95°C, 4 min; 35 x (94°C, 45 s; 56°C, 1 min; 72°C, 1 min); 72°C, 10 min; ∞4°C. | (Chen et al. 2010) |
Búsqueda, descarga y alineamiento de secuencias
Se descargaron secuencias de especies del género Quercus correspondientes a tres códigos de barras de ADN (rbcL, matK e ITS2) disponibles en la base de datos del BOLSYSTEMS (Boldsystems 2021). Se filtraron secuencias de acuerdo con la longitud de nucleótidos documentada para cada marcador y sin bases ambiguas “N”. El alineamiento de las secuencias de referencia y las que se generaron en el laboratorio se llevó a cabo de manera independiente para cada marcador y se ajustaron de forma manual con la herramienta de ClustalW (Thompson et al. 1994) implementado en MEGAX (Tamura et al. 2011).
Análisis de códigos de barras de ADN
Para evaluar la efectividad del poder discriminatorio de los códigos de barras analizados, se utilizaron los métodos de BLAST, distancia genética y mediante la construcción de árboles de unión de vecinos cercanos, con la finalidad de asignar una identidad a las secuencias analizadas, se determinó la frecuencia con la que cada método devolvió la identidad correcta.
Método de identificación con BLAST. Las secuencias de los tres marcadores se concatenaron (por separado) como una base de datos de referencia y mediante la herramienta de BLASTn (NCBI 2021) se realizó un BLAST considerando un valor de E <1 x 10-5 y aciertos máximos de 98 a 100% con una especie (Ross et al. 2008). Se utilizó el método de distancia (más cercana), con la función BestCloseMatch (Meier et al. 2008) del software SPIDER (Brown et al. 2012).
Con base en las secuencias alienadas, el método de unión del vecino más cercano (Saitou y Nei 1987) en MEGA X (Kumar et al. 2018 y Stecher et al. 2020), se evaluó el desempeño discriminatorio mediante el cálculo de la proporción de especies. Las distancias evolutivas se calcularon utilizando el método de 2 parámetros de Kimura (K2P) (Kimura 1980) y están en las unidades del número de sustituciones de bases por sitio. Todas las posiciones ambiguas se eliminaron para cada par de secuencias (opción de eliminación por pares). Únicamente las especies con múltiples individuos que formaron un clado monofilético en el árbol se consideraron identificadas con éxito. Se construyó un árbol de unión de vecinos para cada marcador. El análisis basado en la construcción de árboles proporciona un método conveniente y visualizado que permite evaluar el desempeño discriminatorio mediante el cálculo de la proporción de grupos monofiléticos.
Resultados
En cinco localidades de la región sureste del estado de Coahuila, México (Figura 1), se colectaron e identificaron morfológicamente 11 diferentes especies de encinos del subgénero Quercus, de las cuales siete pertenecen a la sección Quercus y cuatro a la sección Lobatae (Tabla 2). En total se seleccionaron 29 muestras para los análisis genéticos.
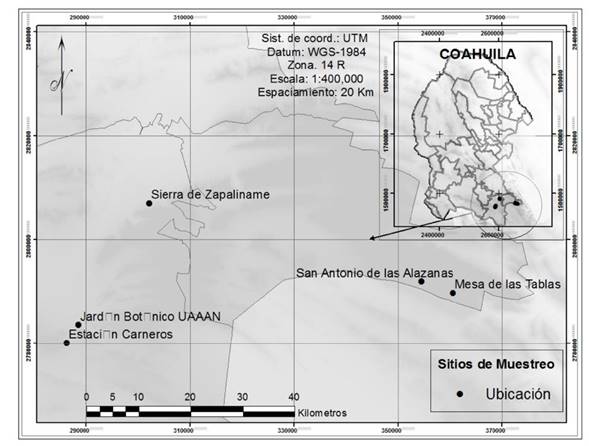
Tabla 2: Especies y localización geográfica de muestras colectadas.
| Especie | Número de muestra | Latitud N | Longitud O | Altitud | Sección |
|---|---|---|---|---|---|
| Quercus microphylla | 1 | 25° 15’ 08.7” | 100° 31’ 14.39” | 2388 | Quercus |
| 2 | 25° 15’ 08.6” | 100° 31’ 14.6” | 2391 | Quercus | |
| 3 | 25° 15’ 09.0” | 100° 31’ 14.4” | 2391 | Quercus | |
| Quecus saltillensis | 1 | 25° 14’ 13.0” | 100° 26’ 39.1” | 2596 | Lobatae |
| 2 | 25° 14’ 13.5” | 100° 26’ 39.3” | 2610 | Lobatae | |
| 3 | 25° 14’ 13.6” | 100° 26’ 39.2” | 2608 | Lobatae | |
| Quercus hintoniorum | 1 | 25° 13’ 03.5” | 100° 23’03.1” | 2864 | Lobatae |
| 2 | 25° 13’ 04.0” | 100° 23’02.6” | 2856 | Lobatae | |
| 3 | 25° 13’ 03.8” | 100° 23’02.8” | 2860 | Lobatae | |
| Quercus pringlei | 1 | 25° 07’ 19.2” | 101° 07’ 12.1” | 2315 | Quercus |
| 2 | 25° 07’ 19.2” | 101° 07’ 13.2” | 2316 | Quercus | |
| 3 | 25° 07’ 17.2” | 101° 07’ 16.4” | 2333 | Quercus | |
| Quercus intricata | 1 | 25° 07’ 16.0” | 101° 07’ 16.6” | 2331 | Quercus |
| 2 | 25° 07’ 16.5” | 101° 07’ 16.3” | 2326 | Quercus | |
| 3 | 25° 07’ 17.0” | 101° 07’ 17.2” | 2333 | Quercus | |
| Quercus laceyi | 1 | 25° 22’ 00.5” | 100° 57’ 58.0” | 1910 | Quercus |
| 2 | 25° 21’ 59.8” | 100° 57’ 58.1” | 1913 | Quercus | |
| 3 | 25° 22’ 01.8” | 100° 57’ 56.3” | 1920 | Quercus | |
| Quercus laeta | 1 | 25° 22’ 02.1” | 100° 57’ 03.6” | 1999 | Quercus |
| 2 | 25° 22’ 01.6” | 100° 57’ 02.4” | 2009 | Quercus | |
| 3 | 25° 22’ 01.1” | 100° 57’01.7” | 2006 | Quercus | |
| Quercus laetaxarizonica | 1 | 25° 22’ 00.4” | 100° 56’ 54.9” | 1982 | Quercus |
| 2 | 25° 21’ 59.9” | 100° 56’ 54.3” | 1994 | Quercus | |
| 3 | 25° 22’ 03.7” | 100° 56’ 50.4” | 1996 | Quercus | |
| Quercus grisea | 1 | 25° 21’ 59.6” | 100° 56’ 53.8” | 2004 | Quercus |
| 2 | 25° 22’ 01.4” | 100° 57’ 00.8” | 1998 | Quercus | |
| 3 | 25° 22’ 04.6” | 100° 56’ 50.2” | 2022 | Quercus | |
| Quercus hipoxantha | 1 | 25° 59’ 07.0” | 101° 28’41.6” | 1478 | Lobatae |
| Quercus gravesii | 1 | 25° 59’ 07.3” | 101° 28’ 41.3” | 1478 | Lobatae |
Se obtuvieron productos de PCR en el 100% de las muestras procesadas, los cuales presentaron el tamaño aproximado esperado en rbcL e ITS2 (600 pb y 400 pb, respectivamente) (Figura 2). En ambos casos la recuperabilidad de las secuencias fue alta (96.5 y 86.2%, respectivamente). Los amplicones obtenidos con el fragmento matK resultaron en secuencias de mala calidad que no se incluyeron en el análisis; por lo cual se analizaron únicamente 64 secuencias de referencia matK que cumplieron con los parámetros de calidad.
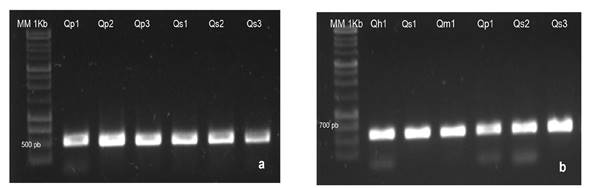
Figura 2: Productos de PCR obtenidos apartir de (a) una región del gen nuclear ITS2 y (b) de una región plastídica del gen rbcL en muestras de ADN de especies de encinos mexicanos.
El análisis incluyó un total de 50 secuencias de consulta 26 del marcador rbcL y 24 del marcador ITS2, provenientes de 11 especies del género Quercus subgénero Quercus, además, se incluyeron 118 especies de referencia (30 rbcL, 27 ITS2 y 61 matK) que provenían de 81 especies del género Quercus (Tabla 3). El conjunto de datos de secuencias del marcador matK incluyó únicamente secuencias de referencia de las regiones de América del Norte y Eurasia. Mientras que los conjuntos de datos rbcL e ITS2 incluyeron secuencias de encinos mexicanos de América del Norte y Eurasia. El análisis involucró diferentes números de secuencias de nucleótidos, por lo que, cada grupo de secuencias tuvo un total diferente de posiciones en el conjunto de datos final de acuerdo con el marcador utilizado. De tal manera que, rbcL generó un total de posiciones de 521 nucleótidos, ITS2 461 nucleótidosy matK 312 nucleótidos.
Tabla 3: Número de accesión de BoIdSystems de secuencias sometidas a comparaciones filogenéticas con tres marcadores.
| Especie | rbcL | matK | ITS2 |
|---|---|---|---|
| Quercus acutissima | GBVG072-11 | ||
| Quercus afares | GBVG075-11 | ITSAJ794-14 | |
| Quercus alba | GBVG078-11 | GBVG080-11 | |
| Quercus aliena | GBVG085-11 | ||
| Quercus alnifolia | GBVG091-11 | ITSAJ1237-14 | |
| Quercus arizonica | GBVG094-11 | ||
| Quercus aucheri | GBVG095-11 | ITSAJ1245-14 | |
| Quercus baloot | GBVG096-11 | ||
| Quercus baroni | ITSAH052-14 | ||
| Quercus berveridifolia | SDH1252-14 | ||
| Quercus bicolor | MKTRT2706-14 | ||
| Quercus brantii | GBVG097-11 | ITSAJ803-14 | |
| Quercus canariensis | GBVG099-11 | ITSAJ921-14 | |
| Quercus castaneifolia | GBVG101-11 | ITSAJ806-14 | |
| Quercus cedrocencis | SDH1254-14 | ||
| Quercus cerris | GBVG115-11 | GBVG102-11 | ITSAJ812-14 |
| Quercus chrysolepsis | GBVS274-13 | ||
| Quercus coccifera | GBVG135-11 | GBVG121-11 | ITSAJ1261-14 |
| Quercus cornelius muelleri | SDH3391-15 | ||
| Quercus corrugata | MHPAC1141-11 | ||
| Quercus crenata | GBVG149-11 | GBVG140-11 | |
| Quercus dentata | GBVG151-11 | ||
| Quercus ellipsodalis | HIMS059-12 | ||
| Quercus engelmanii | SDH3367-15 | ||
| Quercus fabri | GBVG157-11 | ||
| Quercus faginea | GBVG158-11 | ITSAJ959-14 | |
| Quercus floribunda | GBVG160-11 | ITSAJ1295-14 | |
| Quercus frainetto | GBVG161-11 | ITSAJ977-14 | |
| Quercus gambelli | GBVG174-11 | ||
| Quercus garryana | VPSBC1009-13 | GBVG176-11 | |
| Quercus glauca | GBVG185-11 | GBVS270-13 | |
| Quercus griffithii | GBVG187-11 | ||
| Quercus ilex | ATG011-14 | GBVG194-11 | ITSAJ1306-14 |
| Quercus ilicifolia | VASCA016-15 | ||
| Quercus infectoria | API328-14 | GBVG212-11 | |
| Quercus infectoria var. boissieri | ITSAJ912-14 | ||
| Quercus infectoria var. infectoria | ITSAJ1034-14 | ||
| Quercus ithaburensis var. ithaburensis | GBVG218-11 | ||
| Quercus ithaburensis var. macrolepsis | GBVG219-11 | ITSAJ845-14 | |
| Quercus kellogi | SDH1261-14 | ||
| Quercus laeta | GBVG222-11 | ||
| Quercus lanata var. lanata | GBVW3164-13 | ||
| Quercus leucotrichophora | GBVW3167-13 | ||
| Quercus libani | GBVG223-11 | ||
| Quercus lobata | GBVG225-11 | ||
| Quercus lusinatica | GBVG226-11 | ||
| Quercus macranthera | GBVG229-11 | ITSAJ1055-14 | |
| Quercus macrocarpa | FOND165-12 | GBVG233-11 | |
| Quercus mongolica | RINGV016-12 | GBVG237-11 | |
| Quercus montana | GBVG262-11 | ||
| Quercus muhelenbergi | MKTRT534-12 | ||
| Quercus petraea | GBVG295-11 | GBVG278-11 | |
| Quercus petraea var. iberica | GBVG299-11 | ||
| Quercus petrea var. petrea | GBVS1218-13 | ITSAJ1013-14 | |
| Quercus phillyraeoides | GBVG302-11 | ||
| Quercus polycarpa | GBVG307-11 | ||
| Quercus pontica | GBVG308-11 | ITSAJ1121-14 | |
| Quercus prinoides | MKTRT558-12 | ||
| Quercus pseudosemercarpifolia | GBVG309-11 | ||
| Quercus pubescens | GBVG326-11 | GBVG310-11 | ITSAJ1131-14 |
| Quercus pubescens var. pubescens | GBVS1219-13 | ||
| Quercus pyrenaica | GBVG329-11 | ITSAJ1168-14 | |
| Quercus robur | GBVG349-11 | GBVG330-11 | |
| Quercus robur var. robur | GBVG354-11 | ||
| Quercus robur var. pedunculiflora | ITSAJ1074-14 | ||
| Quercus rubra | BPNP195-08 | GBVS271-13 | |
| Quercus semecarpifolia | GBVG372-11 | ||
| Quercus serrata | GBVG373-11 | ||
| Quercus serrata var. brevipetiolata | GBVG385-11 | GBVG384-11 | |
| Quercus shumardii Quercus stewardiana | MKTRT582-12 | GBVX5226-15 | |
| Quercus suber | GBVG404-11 | GBVG390-11 | ITSAJ852-14 |
| Quercus suber x Quercus trojana | ITSAJ891-14 | ||
| Quercus trojana | GBVG408-11 | ITSAJ896-14 | |
| Quercus variabilis | GBVG415-11 | ||
| Quercus vulcanica | GBVG419-11 | ITSAJ1226-14 | |
| Quercus wislizeni var. frutescens | SDH3355-15 | ||
| Quercus wislizeni var. wislizeni | SDH1265-14 | ||
| Quercus wutaishanica | GBVG420-11 | ||
| Quercus x ganderi Quercus x morisii | SDH3343-15 | GBVG422-11 |
Para la identificación con el método de BLAST, se construyeron dos bases de datos con las secuencias de consulta, la primera con 26 secuencias rbcL y la otra con las 24 secuencias ITS2, las cuales fueron alineadas de manera independiente junto con las secuencias de referencia. Los resultados indican que, el método de BLAST proporcionó el porcentaje de identificación más alto con promedios de porcentaje de identidad de 98.1 y 98.3% para los marcadores rbcL e ITS2, respectivamente. Con el método basado en la distancia genética más cercana, los códigos de barras de ADN universal mostraron menos poder discriminatorio, así rbcL, ITS2 y matK tuvieron una tasa de éxito del 3.57, 7.84 y 11.47%, respectivamente.
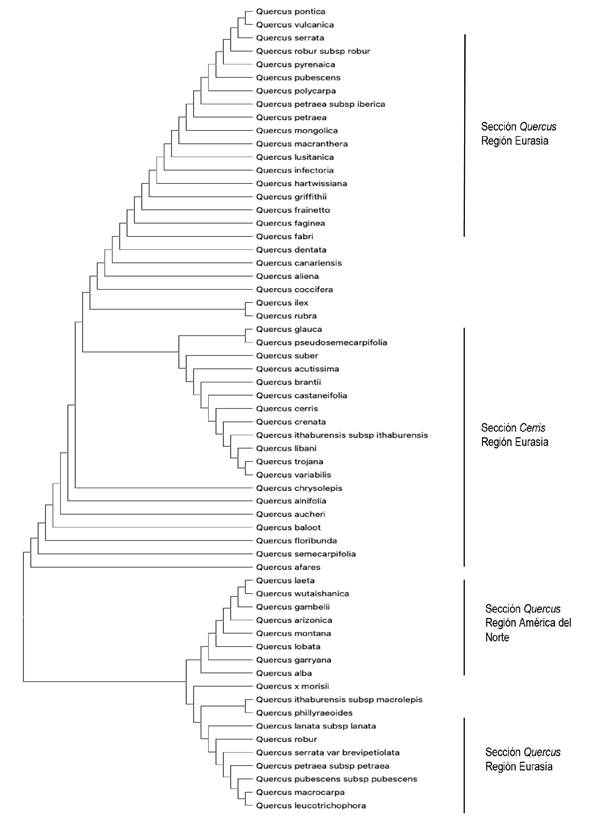
Con base en el método de unión de vecinos cercanos, los taxones de plantas recolectados se agruparon de acuerdo con el análisis de similitud, en cada conjunto de datos se formaron diferentes grupos monofiléticos. En general, en los árboles se aprecian grupos monofiléticos definidos. El porcentaje de discriminación más bajo se generó con el marcador rbcL (64.28%) (Figura 3). Con el marcador ITS2 se logró un porcentaje de discriminación del 80.39%, en la Figura 4 se aprecian algunos grupos monofiléticos. Mientras que, con el marcador matK se obtuvo el porcentaje más alto (81.96%), en la Figura 5 se confirma la separación de las 61 especies por secciones del subgénero y por regiones geográficas, formando tres clados pincipales correspondientes a las secciones Quercus (Eurasia), Cerris (Eurasia) y Quercus (América del Norte). Teniendo en cuenta que en los conjuntos de datos de los marcadores rbcL e ITS2 contenían secuencias de especies mexicanas, ITS2 proporcionó una mejor resolución cladística en este grupo de plantas. Cada árbol generado mostró formas diferentes de acuerdo al agrupamiento de especies estrechamente relacionadas, mientras que las especies lejanamente relacionadas se dispersaron relativamente.

Figura 3: Análisis filogenético molecular usando la región plastídica rbcL para 56 secuencias de nucleótidos con base en la distancia genética K2P
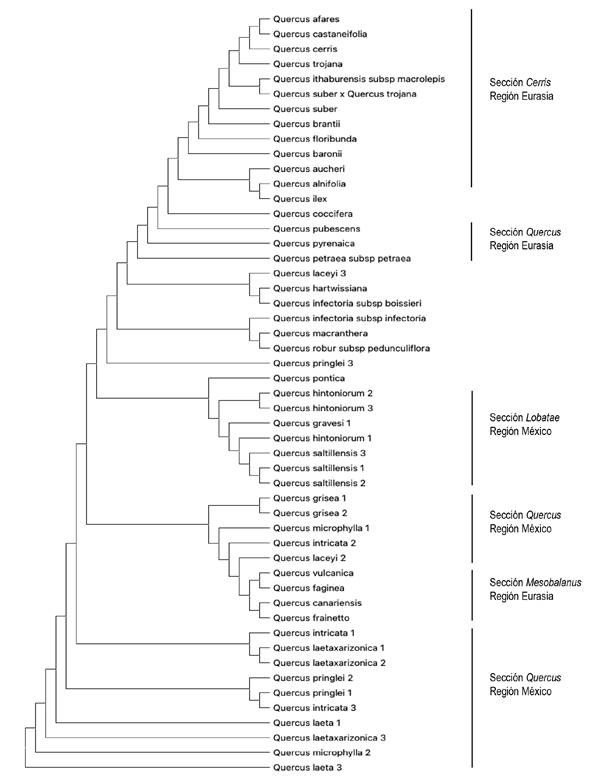
Figura 4: Análisis filogenético molecuar usando la región nuclear ITS2 para 51 secuencias de nucleótidos con base en la distancia genética K2P
Discusión
Las características que un código de barras ideal debería poseer son: cebadores altamente universales y de secuencia corta, pero con suficiente variación entre secuencias que permita la identificación y discriminación entre especies (Erickson et al. 2008, Kress y Erickson 2008, Hollingsworth et al. 2009, Stoeckle et al. 2011). El Grupo de Trabajo CBOL propuso dos regiones del cloroplasto (rbcL y matK), como los códigos de barras centrales, esto, debido a la conservación de sus genes y a la universalidad de amplificación de los cebadores utilizados (De Groot et al. 2011, Pang et al. 2012 y Saarela et al. 2013). Los códigos de barras seleccionados en este estudio presentaron resultados variables. La secuencia rbcL mostró una cobertura alta de amplificación (100%) y secuenciación (96.5%) en las once especies de encinos mexicanos, resultados similares han sido reportados en diferentes grupos de plantas de especies arbóreas, tropicales y medicinales (Gonzalez et al. 2009, Parmentier et al. 2013, Kang et al. 2017, Maloukh et al. 2017). De igual manera en encinos italianos, se obtuvieron resultados positivos en la amplificación del ADN (Piredda et al. 2011). Lo anterior, confirma que es posible la amplificación y secuenciación de buena calidad, en condiciones estándar y en una amplia gama de taxones concebadores rbcL utilizados en este estudio. Por otro lado, el bajo éxito de amplificación y secuenciación utilizando cebadores matK se ha informado en diferentes grupos de plantas, en angiospermas y gimnospermas, incluso en grupos de plantas específicos (Kress y Erickson 2008, Bafeel et al. 2011, Maloukh et al. 2017, Amandita et al. 2019). Mientras que Piredda et al. (2011), al eliminar de sus análisis los datos de secuencias provenientes de la región matK (universal) sugerida por el CBOL en encinos italianos, debido a la baja amplificación por PCR, incluso con cebadores específicos para dicho taxón no obtuvieron buenos resultados de amplificación y su poder discriminatorio fue relativamente bajo. Mientras que Yang et al. (2016) reportaron una alta tasa de amplificación con cebadores diseñados para el grupo de plantas de Quercus de la región de China, pero con bajo rendimiento en la identificación de especies. Resultados alentadores se han logrado en algunos grupos de plantas específicos como Asteraceae (Gao et al. 2010), Lamiaceae (De Mattia et al. 2011), Tetrastigma (Fu et al. 2011) y en palmas (Jeanson et al. 2011). La variabilidad de resultados obtenidos con esta región, puede deberse a la diversa tasa evolutiva en diferentes taxones, ya que la región matK tiene una alta tasa de sustituciones de nucleótidos que puede estar influenciada por factores climáticos que afecta la conservación del locus de diferentes grupos de plantas (Hilu et al. 2003, Gillman et al. 2010). La utilidad de la región matK como código de barras de ADN es fundamental en la discriminación e identificación de especies, por lo que, es imprescindible continuar con la búsqueda de cebadores que permitan la amplificación de ADN de grupos taxonómicos difíciles o grupos de plantas específicos (Hollingsworth et al. 2011, Piredda et al. 2011). En 2011 el CBOL propuso la región ITS como un código de barras potencialmente útil para la identificación de especies de plantas (China Plant BOL Group et al. 2011). Su utilidad ha sido respaldada en diferentes investigaciones que incluyen una amplia gama de grupos taxonómicos donde se han logrado amplificaciones exitosas y secuencias de alta calidad (Bellarosa et al. 2005, Chen et al. 2010, Pang et al. 2010, Simeone et al. 2013, Chen et al. 2015). Aúnque los resultados de amplificación y secuenciación pueden ser variables según el taxón de plantas (Kang et al. 2017). En este estudio los cebadores ITS2 presentaron una tasa de amplificación del 100% y permitieron obtener secuencias de alta calidad en un 86.2% de las muestras analizadas. La característica principal de la región ITS2 es que es una secuencia corta, lo cual permite una alta tasa de amplificación, además, sus estructuras secundarias se conservan y proporcionan información útil en biología (Meyer y Paulay 2005 y Chen et al. 2010). Sin embargo, a pesar del desarrollo de la tecnología de los códigos de barras de ADN, aún no se cuenta con un código estándar para la identificación de taxones de plantas (Ballardini et al. 2013, Hollingsworth et al. 2011).
Uno de los propósitos principales de la tecnología de códigos de barras de ADN es la identificación de especies desconocidas, de tal manera que, una secuencia de código de barras particular coincida con secuencias confirmadas disponibles en las bases de datos. Al respecto los tres métodos de análisis utilizados proporcionaron diferentes tasas de discriminación en las especies de encinos del subgénero Quercus. Teniendo en cuenta que, los conjuntos de datos de los marcadores rbcL e ITS2 contenían secuencias de consulta de especies mexicanas, el número de las coincidencias correctas en BLAST proporcionó el poder de identificación alto con 98.1 y 98.3%, respectivamente. De igual manera, en un grupo de encinos de origen chino y en un grupo taxonómico específico (Apocynaceae) se encontró que el método de BLAST mostró la identificación más alta, respecto a otros métodos de identificación a nivel de género y especie (Cabellin et al. 2016, Yang et al. 2016).
El método basado en la distancia genética mostró tasas de discriminación extremadamente bajas para las especies de encinos 3.57% con rbcL, 7.84% con ITS2 y 11.47% con matK. Resultados similares obtuvieron Pang et al. (2019) en donde, la distancia genética mostró las tasas de discriminación más bajas para las especies de encinos italianos con porcentajes que van del 12.50% para rbcL y 25% para el marcador matK. Por otro lado, en especies de encino chino el método basado en la distancia genética también mostró tasas de discriminación bajas (0 al 17.14%) (Pang et al. 2010). En contraste, Chen et al. (2010) obtuvieron resultados favorables cuando utilizaron el método de distancia genética más cercana, aplicado a especies de plantas medicinales con el marcador ITS2, el cual identificó correctamente el 90.3 y el 99.7% de las muestras a nivel de especie y género, respectivamente. En este estudio la aplicación de este método parece poco práctico en la codificación de especies de encinos dado que las distancias genéticas intra e interespecíficas se superponen en gran medida. El análisis basado en la distancia genética ha sido propuesto como un método altamente sensible a procesos de divergencia reciente entre especies, lo que puede resultar en una clasificación de linajes incompletos, y afectar el resultado obtenido aplicando dicho método (Simeone et al. 2013). En el caso específico de encinos diferentes autores mencionan la baja identificación basada en la distancia genética, debido a que, un muestreo mayor puede conducir a distancias genéticas superpuestas, además de la frecuente hibridación entre especies y una baja tasa de variación (Piredda et al. 2011, Simeone et al. 2013).
Junto con los datos moleculares de los códigos de barras de ADN, la construcción de árboles se utilizó para probar las relaciones filogenéticas de las cinco secciones del subgénero Quercus basadas en la morfología, además de probar la separación de grupos por regiones geográficas. Mediante la metodología de unión de vecinos cercanos y distancias evolutivas para 92 especies de encinos se generaron clados individuales de acuerdo al marcador utilizado. Los resultados de los árboles proporcionaron información importante sobre las relaciones filogenéticas de especies de encinos. Este método mostró tasas de discriminación relativamente altas. Los encinos (Quercus L.) pertenecen a un grupo de plantas con alta complejidad taxonómica, actualmente reconocidos dos subgéneros: Quercus distribuido principalmente en América del Norte y Eurasia (región del Mediterráneo) y el subgénero Cyclobalanopsis distribuido principalmente en el sudeste asiático. En este estudio, los árboles filogenéticos confirmaron la separación de grupos monofiléticos de acuerdo con datos morfológicos (Valencia 2004, Oh y Manos 2008), en secciones del grupo Quercus (Cerris, Lobatae, Mesobalanus, Protobalanus y Quercus) con el uso de los marcadores ITS2 y matK los cuales generaron un poder de discriminación del 80.39 y 81.96%, respectivamente (Figuras 4 y 5). Resultados similares se han obtenido en diferentes grupos de plantas (Amandita et al. 2019, Chen et al. 2010). La variación en el poder de identificación se debe probablemente a la diferenciación en las tasas de mutación y evolución que resulta en una menor información genética de los marcadores (Palmer et al. 2000, Yang et al. 2017). Con lo anterior, se confirma que la inclusión de la región ITS2 como código de barras de ADN parece ser esencial en la discriminación de grupos de plantas estrechamente relacionados (Chen et al. 2010, Simeone et al. 2013). Además, en el árbol generado con secuencias del marcador matK se observa una separación de especies de acuerdo a su centro de diversificación, americano o euroasiático (Figura 5), lo anterior, respalda la posibilidad que las especies de encinos provienen de un doble origen (América y Eurasia) (Oh y Manos 2008, Hubert et al. 2014, Yang et al. 2017). Por otro lado, el marcador rbcL indicó limitaciones en su poder de identificación, con un porcentaje bajo (64.28%), lo cual condujo a un árbol poco claro en la generación de grupos monofiléticos (Figura 3). Este resultado puede deberse al hecho de que, a pesar de que la región rbcL tiene una amplificación casi universal, su tasa de evolución es relativamente baja (Simeone et al. 2013).
México destaca como un país con una diversificación excepcional de encinos asociado a una mayor heterogeneidad ecológica. La disponibilidad de humedad en los nichos ecológicos, afecta directamente las tasas de evolución y diversificación de linajes de encinos en México, en comparación con la región de América del Norte (Torres et al. 2011 y Hipp et al. 2018). Finalmente, la comparación de marcadores rbcL e ITS2 respecto a secuencias de encinos de origen mexicano permite inferir que el marcador ITS2 proporciona una mejor identificación y agrupamiento de estas especies de acuerdo con datos morfológicos (Valencia 2004, Villarreal et al. 2008).
Conclusiones
Con base en los resultados obtenidos con la metodología de unión de vecinos cercanos, altos porcentajes de amplificación y recuperabilidad de secuencias, es concluyente que el marcador de la región nuclear ITS2 funciona razonablemente bien para la identificación especies de encinos. De igual manera la región matK tuvo un buen desempeño de acuerdo a la discriminación de especies obtenida, pero su universalidad de amplificación en un amplio grupo de plantas aún es cuestionable. Mientras que el marcador rbcL es ampliamente utilizado por sus características de amplificación y obtención de secuencias, su baja tasa evolutiva no permite un uso eficiente de dicho marcador en la identificación de especies. Es importante mencionar que, según datos del BOLSYSTEMS, únicamente alrededor del 5% de especies de encinos mexicanos cuentan con al menos un código de barras de ADN, lo que muestra la importancia de continuar y expandir los estudios de esta índole para las especies de encinos de México.

