











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkTheobroma cacao L. (Malvaceae) es una especie vegetal de naturaleza tropical que proporciona, dentro de un ámbito sostenible, beneficios económicos y ambientales precedidos por un aprovechamiento resiliente por parte de más de seis millones de agricultores en el mundo. Debido a lo anterior, el “árbol del chocolate” posee una importancia económica y biocultural sumamente importante (Ricaño-Rodríguez et al. 2018). Las regiones productoras de cacao se centran en gran medida en las proximidades de 13 de las regiones con mayor diversidad biológica del planeta (Motamayor et al. 2002). Dado su origen en la cuenca amazónica y la distribución de especies del género Theobroma a lo largo del continente americano, hasta la fecha se han caracterizado al menos tres variedades genéticas (i.e. cacao Criollo, Forastero y Triniatario) delimitadas por su fenología y morfología respectivamente.
Las poblaciones de T. cacao situadas en el sureste de México poseen al menos 12 alelos claramente diferenciados entre especies y cabe mencionar que el Estado de Tabasco es el principal productor de cacao en la República. En principio, el cacao Criollo que es considerado una variedad ancestral, supondría un parteaguas en la historia evolutiva de todas las variedades conocidas (Motamayor et al. 2002). Gracias a diversos estudios biogeográficos y de genética poblacional, se ha reagrupado gran parte del germoplasma de cacao en el mundo en 10 clústeres genéticos representativos (i.e. Marañón, Curaray, Criollo, Iquitos, Nanay, Contamana, Amelonado, Purús y Nacional y Guiana) (Motamayor et al. 2008). Es importante mencionar que el estudio de la diversidad genética de esta especie se ha realizado principalmente con herramientas de diagnóstico molecular (Santana et al. 2016).
En este sentido, la mayoría de las indagaciones relacionadas con el uso de recursos biotecnológicos dirigidos al estudio del cacao, tienen como objetivo principal; dilucidar las diferencias entre variedades criollas y forasteras. Ahora bien, existe una herramienta de diagnóstico molecular relativamente novedosa denominada “genotipado por secuenciación” (GBS; Genotyping-by-Sequencing) que se ha desarrollado bajo un enfoque de análisis rápido y robusto, que simplifica el flujo de datos de muestras de ADN multiplexadas, combinando la caracterización de marcadores genéticos a lo largo del genoma previa reducción secuencial mediante enzimas de restricción (RE) (Poland & Rife 2012). La flexibilidad, rapidez y robustés aunadas al bajo costo del GBS hacen de éste, una herramienta excelente para muchas aplicaciones biotecnológicas en los campos de la fitogenética y fitomejoramiento, entre otros.
Así, el genotipado por secuenciación suele ser simple, rápido, extremadamente específico y reproducible, además de que permite caracterizar regiones genómicas prácticamente inaccesibles para la mayoría de los métodos de secuenciación (Elshire et al. 2011). Las variaciones alélicas del genoma de una especie se estudian también a través de sus polimorfismos de un solo nucleótido (SNP; Single Nucleotide Polymorphism) (Wang et al. 1998). Se sabe que este tipo de características genéticas representa diferencias fenotípicas que se rigen por menos del 0.1 % de las diferencias moleculares restantes entre individuos (Weber & May 1989).
Debido a que existen relativamente pocas regiones con SNPs identificadas en el genoma del cacao y, dada la naturaleza bialélica de la especie (Rafalski 2002, Kuhn et al. 2008, Lima et al. 2009, Livingstone et al. 2011), es imperativo caracterizar un mayor número de loci que proporcionen datos específicos sobre la historia evolutiva, naturaleza de regiones codificantes y transcripción de péptidos ribosomales del género Theobroma. En virtud de ello, el proyecto de secuenciación del genoma del cacao Criollo versión 2 (http://cocoa-genome-hub.southgreen.fr) (Argout et al. 2017) al igual que otros proyectos dedicados a la búsqueda de SNPs y regiones conservadas, enriquecen los repositorios correspondientes con nuevos datos moleculares generados mediante secuenciaciones de nueva generación (NGS; New Generation Sequencing) las cuales logran identificar variaciones alélicas únicas.
Lo anterior técnicamente se traduce en el diseño de herramientas biotecnológicas capaces de soslayar parte de las necesidades agrarias de los productores de cacao (e.g. selección adecuada de variedades: (1) mayormente productivas y (2) resilientes al medio ambiente, así como (3) con mayor resistencia a plagas fitopatógenas) (Livingstone et al. 2012). A la luz de las consideraciones anteriores, el objetivo principal de este trabajo fue analizar parte de la naturaleza polimórfica de nucleótidos únicos de loci de variedades nativas mexicanas de cacao provenientes del Estado de Tabasco, a partir de reducciones genómicas (escisiones de ADN con endonucleasas específicas) que generan fragmentos restrictivos.
Así, los resultados de este trabajo se traducirían a futuro en la utilización de herramientas biotecnológicas que promuevan un mejor aprovechamiento del cacao en un sentido resiliente y biocultural, pero sobre todo, económicamente sostenible.
Materiales y métodos
Obtención de material biológico. Las regiones cacaoteras de las cuales proviene el material biológico empleado en este estudio, se encuentran situadas en las inmediaciones rurales de los municipios de Cárdenas, Huimanguillo, Comalcalco, Paraíso, Jalpa de Méndez y Cunduacán Tabasco, así como los municipios de Ixtacomitán y Pichucalco, Chiapas (Figura 1). Dichas zonas de muestreo son representativas para la región productora de cacao de ambos Estados. Se seleccionaron 20 individuos de variedades nativas de cacao de edad adulta que forman parte del sistema agroforestal de parcelas productoras. A cada variedad se le asignó una clave de identificación correspondiente (i.e. muestra; cuya etiqueta se compone de las primeras letras de los nombres y apellidos de los productores, así como el municipio del que provienen y el número de planta correspondiente). Los criterios de selección de los individuos fueron: mayor índice de robustés y número de frutos, así como buena condición fitosanitaria y longevidad. Posteriormente, en la finca experimental “Los Chocos” se injertaron varetas de los árboles seleccionados en tutores de cacao que germinaron previamente a partir de semillas. Una vez que los 20 injertos desarrollaron hojas jóvenes se colectaron muestras foliares, almacenándolas en frío hasta su traslado al laboratorio. Las hojas recolectadas se almacenaron a 4 ºC hasta su posterior desinfección con NaOCl al 0.02 % y maceración en frío con nitrógeno líquido. El material biológico pulverizado se almacenó a -20 ºC hasta su uso.

Figura 1 Ubicación geográfica de las parcelas agroforestales de las cuales provienen las plantas de cacao empleadas en este estudio. La georreferenciación fue generada con el software QGIS ver. 3.6.3.
Extracción de material genético. Se partió de 200 mg de tejido en polvo adicionando 900 µL de buffer de Lin (Lin et al. 2001), 12 µL de β-mercaptoetanol y 2 µL de RNAsa (20 µg/mL). Se mezcló perfectamente y se incubó a 37 °C durante 1 h y 600 rpm, incorporando un volumen de mezcla de fenol-cloroformo-alcohol isoamílico (25:24:1). Se centrifugó a 12,000 rpm/10 min a 4 °C (dos veces). Posteriormente se adicionó un volumen (equivalente a la mezcla de reacción anterior) de isopropanol frío al 100 % y se incubó a -20 °C durante 1 h, seguido de una centrifugación a 12,000 rpm/10 min a 4 °C y se descartó el sobrenadante. A la pastilla restante se le adicionaron 500 µL de etanol al 70 % y una posterior centrifugación a 12,000 rpm/30 s. Se eliminó el etanol dejando secar el remanente a temperatura ambiente. Por último, se hidrató la pastilla con 30 µL de agua libre de nucleasas (AMBION), se cuantificó por espectrofotometría A260/A280 y se corroboró la integridad del ADN mediante electroforesis en gel de agarosa. El material genético se almacenó a -70 ºC hasta su uso.
Selección de enzimas de restricción y diseño de adaptadores. La digestión previa del material genético se realizó con las enzimas de restricción Bgl II y Ddel I que generaron extremos cohesivos. De manera complementaria, se utilizaron dos adaptadores con sus respectivas secuencias cortas de ADN (i.e. barcodes; cada adaptador termina con una secuencia de 4 a 8 pb en el extremo superior 3´ de la cadena nucleotídica y 3 pb en el extremo 5´ de la hebra inferior, que es complementaria al extremo cohesivo generado por Bgl II y Ddel I (Figura 2). Las secuencias nucleotídicas que comprenden los adaptadores correspondientes son las siguientes: 5´-ACACTCTTTCCCTACACACGACGCTCTTCCGATCxxxx y 5´-TNAAGACTGGAAGAGCACACGTCTGAACTCCAGTCAyyy en donde “xxxx” y “yyy” representan complementos secuenciales. El segundo adaptador posee extremos cohesivos compatibles únicamente con las enzimas de restricción: 5´-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTNA y 5´-GATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT.

Figura 2 Esquema de formación del banco GBS. “xxxx” y “yyy” representan los complementos secuenciales de los mismos. El segundo adaptador posee extremos cohesivos compatibles únicamente con las enzimas de restricción.
Generación de bibliotecas genómicas (genomas reducidos) y secuenciación por síntesis. Cada banco de genes (genotecas) se generó siguiendo la metodología propuesta por Elshire et al. (2011) con algunas modificaciones. Se construyeron 20 librerías con una concentración de ADN de entre 6.04 y 52.2 ng/µL. Los oligonucleótidos que comprenden los extremos de cada adaptador fueron diluidos por separado en buffer TE (50 µM) y anillados por termociclación (desnaturalización 95 ºC, 2 min; anillamiento y extensión (ramp down) hasta 25 ºC (0.1 ºC/s), 30 min; enfriamiento (hold) 4 ºC). La concentración de adaptadores se cuantificó por fluorimetría.
Las muestras de ADN que contaban con los adaptadores ligados fueron digeridas enzimáticamente (4 U de Bgl II y Ddel I respectivamente) en 20 µL de buffer NED 1 X mediante incubación a 75 ºC por un periodo de 2 h. Los adaptadores se ligaron a los extremos cohesivos adicionando 30 µL de una solución 1.66 X de buffer ligasa. Las muestras se incubaron a 22 ºC × 1 h y posteriormente se calentaron a 65 ºC × 30 min. Cada muestra de ADN digerido se purificó utilizando un kit comercial (QIAquick PCR Purification Kit; Qiagen, Valencia, CA) siguiendo las instrucciones del fabricante. El material genómico se diluyó en agua ultra pura en un volumen final de 50 µL.
Los fragmentos de restricción de cada biblioteca fueron amplificados en mezclas de reacción que contenían 2 µL de ADN plantilla, 25 µL 1 X HotStarTaq Master Mix Kit (QIAGEN, Valencia, CA) y 20 pmol por separado de los siguientes primers: (D501) 5´-ATGATACGGCGACCAGATCTAC-ACTATAGCCTACACTCTTTCCCTACACGACG y (D701) 5´-CGTGTCGACACTTGAGGT-CAGTGTAATGAGC. Los parámetros de amplificación fueron los siguientes: 72 ºC × 5 min, 98 ºC × 30 s, seguido de 18 ciclos a 98 ºC × 30 s, 65 ºC × 30 s, 72 ºC × 30s y un paso de extensión final a 72 ºC × 5 min. Las bibliotecas consideradas aptas para secuenciación contenían amplicones de ≈ 450-500 pb de longitud. El proceso de secuenciación se realizó en una plataforma NextSeq 500 (Illumina) con una profundidad de lectura de 1×150 (número de veces que una región diana es secuenciada o leída), considerando como mínimo 4,5 millones de lecturas por muestra.
Análisis de datos mediante filtrado en crudo y alineación de secuencias reducidas. Las secuencias genómicas se filtraron bajo los siguientes criterios: (1) alineación idéntica con los adaptadores y los extremos nucleotídicos remanentes del sitio de corte de las enzimas Bgl II y Ddel I, respectivamente; (2) ausencia de dímeros en los adaptadores; (3) ausencia de “Ns” (insertos de ADN) en las primeras 100 pb de cada lectura. De esta manera se generaron archivos crudos (.qseq) con instrucciones secuenciales. Posteriormente, se construyó una base de datos que incluyó secuencias derivadas de la digestión in silico con las enzimas de restricción antes mencionadas usando como referencia el genoma del cacao Criollo (B97-61/B2 versión 2; https://cocoa-genome-hub.southgreen.fr/blast) (GenBank assembly accession: GCA_000208745.2) (Argout et al. 2017).
Se mantuvieron las lecturas con un valor Q = 10 (las puntuaciones Q se definen como una propiedad logarítmicamente relacionada con la probabilidad de error de la base exponencial; Q = -10 log10P: Phred score: 1 error cada mil bases secuenciadas) con la finalidad de maximizar el número de lecturas secuenciales útiles en el análisis bioinformático. Una vez filtrados los datos, cada secuencia se alineó con el genoma de referencia utilizando la herramienta Burrows-Wheeler aligment tool (BWA) (Li & Durbin 2009). Igualmente se empleó el algoritmo BLASTn con un corte de valor esperado E = 1e-2 (Herramienta de Búsqueda de Alineaciones Básicas Locales; https://blast.ncbi.nlm.nih.gov/Blast.cgi) para analizar lecturas que no hubiesen alineado con BWA, tomando como referencia la base de datos del repositorio de nucleótidos del NCBI (Centro Nacional para la Información Biotecnológica; https://www.ncbi.nlm.nih.gov).
Análisis descriptivo de polimorfismos de nucléotidos únicos. Para analizar estadísticamente las secuencias genómicas reducidas filtradas y caracterizar los posibles SNPs presentes en las muestras de cacao, se utilizó el software GenAIEx 6.2 (Peakall & Smouse 2006, 2012). Para cada muestra se calculó la heterocigosidad observada y esperada, frecuencia alélica, número total de clústeres, número de loci, parálogos filtrados, así como número y frecuencia de sitios polimórficos.
Análisis de evolución molecular taxonómica. El estudio de la historia evolutiva del banco de genes se basó en tres métodos distintos: (1) unión de secuencias vecinas (Neighbor-Joining) (Saitou & Nei 1987); (2) agrupamiento jerárquico aglomerado simple con media aritmética (UPGMA; Unweighted Pair Group Method with Arithmetic Mean) (Sneath & Sokal 1973), en ambos casos se computaron 500 repeticiones (Felsenstein 1985) calculando las distancias evolutivas con el método de probabilidad máxima compuesta (Tamura et al. 2004) y eliminando todas las posiciones en las secuencias que contenían brechas (gaps) y datos vacíos; (3) máxima verosimilitud (MLE; Maximum Likelihood Estimation) (Tamura & Nei 1993). Cada análisis evolutivo se realizó mediante el software MEGA7 versión 7.0 (Kumar et al. 2016) y se generaron árboles filogenéticos óptimos, complementados por análisis BLASTn contra el genoma de referencia del cacao.
Caracterización de regiones codificantes y evolución mínima de transcritos proteicos. Para identificar regiones codificantes (CDs) en las secuencias de nucleótidos de las muestras de cacao, se realizó un análisis BLASTn tomando como referencia CDs previamente predichos en el genoma del cacao Criollo. Las regiones de los loci que mostraron mayor porcentaje de homología fueron analizadas estadísticamente respecto a su longitud de coincidencia y posición inicial y final de ensamblado. La predicción funcional de transcritos de las regiones codificantes se llevó a cabo con secuencias de aminoácidos respectivamente traducidos. De manera complementaria se realizó una predicción ontológica utilizando como referencia anotaciones funcionales proteicas provenientes del repositorio UniProtKB (https://uniprot.org).
Las alineaciones secuenciales se realizaron con ayuda de los algoritmos RPS-BLAST, BLASTP y PHI-BLAST (Constraint-based Multiple Alignment Tool) (Papadopoulos & Agarwala 2007). Con las secuencias de aminoácidos anteriores se generó un análisis filogenético empleando el método de evolución mínima propuesto por Rzhetsky & Nei (1992), complementado por una prueba de arranque de 500 repeticiones. Las distancias evolutivas se calcularon con el método de número de diferencias de aminoácidos por secuencia (Nei & Kumar 2000) y un análisis de intercambio de secuencias vecinas (Close-Neighbor-Interchage; CNI). Previamente se generó un árbol primario (Saitou & Nei 1987) y se eliminaron todas las posiciones con huecos (gaps) y datos faltantes. Se tomó un conjunto total de datos finales de 321 posiciones y el análisis evolutivo se realizó con el software MEGA7 versión 7.0 (Kumar et al. 2016).
Resultados
Análisis de secuencias crudas. El análisis de calidad de los datos crudos generó un banco de datos para cada genoteca (análisis fastQC; control de calidad) con un valor mínimo de 6×106 y máximo de 9×106 lecturas en un rango de 0 a 150 pb. Mediante el pipeline bioinformático se filtraron las muestras dependiendo de la calidad de sus lecturas (i.e. 62,686,733 lecturas que pasaron el filtrado de calidad; 471,551 lecturas retenidas que se cortaron debido a la detección de adaptadores; 63,159,284 lecturas que mantuvieron una longitud suficiente) (Figura 3). El resultado de la formación de clústeres sugirió un total de 984,017 reagrupaciones con profundidad promedio de 44,867 σ 138.56. Asimismo, la tasa de error a priori (H) y heterocigosidad (E) entre las muestras mostraron valores < 0.02 y 0.001 respectivamente.

Figura 3 Frecuencias de polimorfismos (SNPs) por muestra y número de lecturas realizadas en el análisis GBS. La frecuencia de polimorfismos se expresa en porcentajes totales encontrados en los genomas reducidos (interior de las barras). La cantidad de lecturas por secuenciación* se encuentra fuera de las barras. El análisis de calidad de los datos generó un biobanco para cada genoteca.
Ensamblado genómico. Se determinó el número de loci, número de loci con cobertura de profundidad y filtro de parálogos > 6, número de sitios en loci y la frecuencia de sitios polimórficos para cada una de las muestras (Tabla 1). La frecuencia de polimorfismos por muestra se observa en la Figura 3 y el porcentaje de SNPs sugiere un rango del 2 al 11 % con un promedio de 5 %. La longitud consenso se conformó por 1,824,218 bases que representaron el 0.54 % del tamaño del genoma a una profundidad mínima de 6×.
Tabla 1 Estadística de ensamblados de genomas reducidos de cacao.
| Muestra | No. de loci | No. de loci con cobertura > 6 | No. de loci con parálogos > 6 | No. de sitios en loci | No. de sitios polimórficos | Frecuencia de sitios polimórficos |
|---|---|---|---|---|---|---|
| MTJ01 | 47,698 | 18,377 | 15,964 | 2,296,477 | 1,543 | 0.0006719 |
| ACP03 | 47,531 | 19,640 | 16,656 | 2,399,369 | 8,415 | 0.0035072 |
| JGH02 | 46,038 | 21,408 | 18,611 | 2,622,853 | 1,723 | 0.0006569 |
| MTJ03 | 47,627 | 19,286 | 16,587 | 2,393,255 | 5,051 | 0.0021105 |
| JGH01 | 43,654 | 16,751 | 14,451 | 2,087,304 | 2,925 | 0.0014013 |
| EJP01 | 51,066 | 22,653 | 19,407 | 2,757,215 | 8,393 | 0.0030440 |
| LlHC02 | 44,332 | 17,311 | 14,968 | 2,162,333 | 2,965 | 0.0013712 |
| ACP01 | 48,517 | 17,780 | 15,141 | 2,185,854 | 7,180 | 0.0032848 |
| MAJ02 | 45,612 | 18,092 | 15,633 | 2,236,883 | 1,661 | 0.0007426 |
| VAP02 | 50,693 | 17,320 | 14,926 | 2,147,756 | 2,736 | 0.0012739 |
| CCC03 | 42,645 | 16,135 | 14,032 | 2,026,246 | 1,239 | 0.0006115 |
| ALC03 | 51,271 | 17,187 | 14,839 | 2,138,410 | 1,720 | 0.0008043 |
| EJP03 | 47,146 | 16,389 | 14,141 | 2,047,286 | 3,846 | 0.0018786 |
| ALC02 | 44,520 | 17,003 | 14,669 | 2,117,500 | 2,374 | 0.0011211 |
| MAJ01 | 46,614 | 19,131 | 16,248 | 2,429,651 | 4,696 | 0.0020158 |
| VAP03 | 45,738 | 18,268 | 15,632 | 2,240,065 | 5,633 | 0.0025147 |
| GAI02 | 51,467 | 19,717 | 16,839 | 2,418,981 | 7,497 | 0.0030992 |
| MAJ03 | 44,642 | 17,746 | 15,438 | 2,229,200 | 2,913 | 0.0013067 |
| GAI01 | 52,818 | 20,787 | 17,691 | 2,532,699 | 9,933 | 0.0039219 |
| LlHC03 | 48,388 | 19,913 | 16,936 | 24,27,152 | 9,708 | 0.0039998 |
Evolución molecular taxonómica. Los resultados de los análisis evolutivos moleculares de los genomas reducidos de cacao se muestran de manera detallada en la Figura 4 (Neighbor-Joining), Figura 5 (UPGMA) y Figura 6 (MLE) respectivamente. La suma total de la longitud de las ramas (sustituciones por sitio, incluyendo las distancias evolutivas para cada una de éstas y tomando en cuenta su probabilidad máxima compuesta eliminando todas las posiciones que contenían espacios faltantes), para cada caso fueron las siguientes: Neighbor-Joining = 0.00728643; UPGMA = 0.00725912. En ambos análisis hubo un total de 1,824,218 posiciones en el conjunto de datos finales. Respecto al análisis MLE la probabilidad de registro más alta fue de -24,308,138.44 sustituciones. El conjunto de resultados de divergencia evolutiva se calculó con estimadores mínimos para cada caso y en virtud de ello, se observa una reagrupación distinta de muestras. Respecto a la filogenia evolutiva obtenida con los métodos de unión de secuencias vecinas (Figura 4) y agrupamiento jerárquico aglomerado (Figura 5) no es posible distinguir del todo un taxón con clústeres totalmente diferenciados, pues en principio las muestras sugieren una jerarquía que se organiza ya sea por pares o subramas primarias dependiendo de los valores obtenidos de los parámetros de re-muestreo replicado (bootstrapping).

Figura 4 Árbol filogenético Neighbor-Joining (NJ) (Saitou & Nei 1987). Se muestra el árbol óptimo con la suma de longitud de rama = 0.00728463. El porcentaje de árboles replicados en los taxones asociados agrupados en la prueba de arranque (500 repeticiones) se muestra junto a las ramas (Felsenstein 1985). El árbol se encuentra a escala con longitudes de rama en las mismas unidades que las distancias evolutivas utilizadas para inferir el árbol filogenético. Las distancias evolutivas se calcularon utilizando el método de probabilidad máxima compuesta (Tamura et al. 2004) y se encuentra en unidades del número de sustituciones de base por sitio. Se observó un total de 1,824,218 posiciones en el conjunto de datos finales. Los análisis evolutivos se realizaron con MEGA7 versión 7.0 (Kumar et al. 2016).
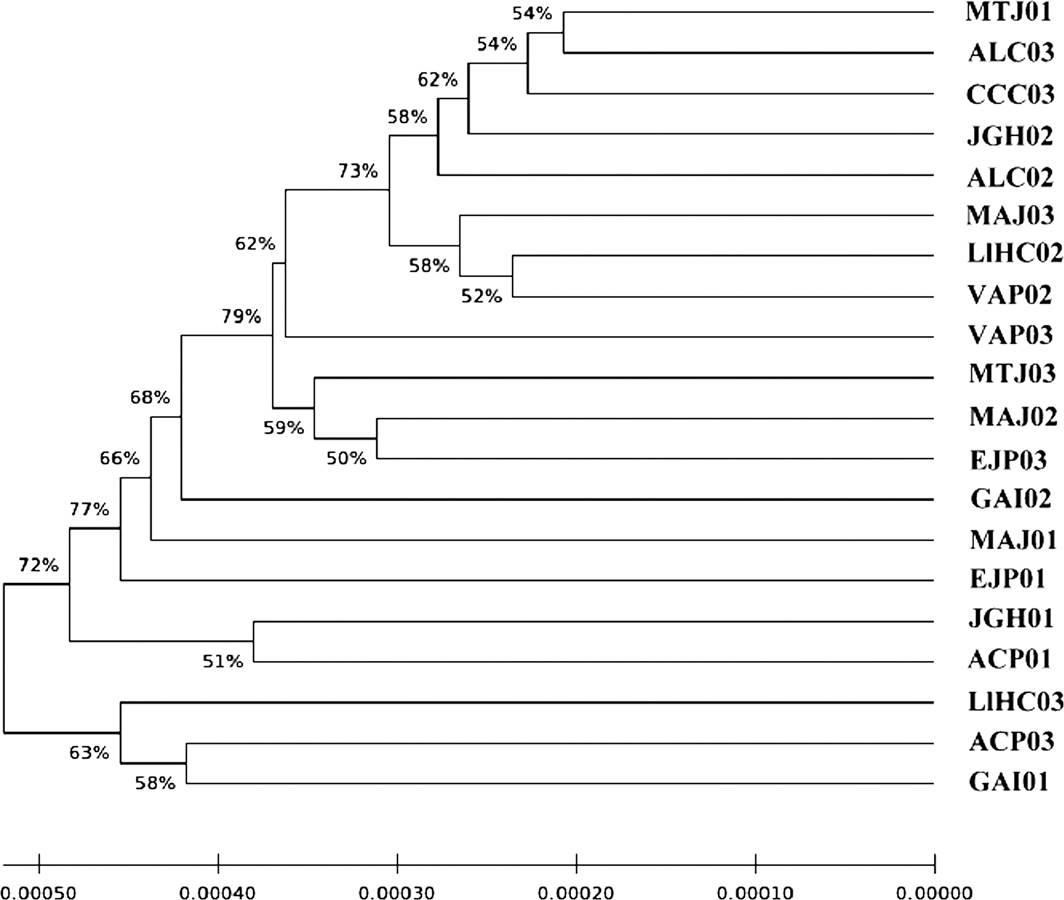
Figura 5 Árbol filogenético UPGMA (Sneath & Sokal 1973). Se muestra el árbol óptimo con la suma de la longitud de rama = 0.00725912. El porcentaje de árboles replicados en los taxones asociados, agrupados en la prueba de arranque (500 repeticiones) se muestran junto a las ramas (Felsenstein 1985). El árbol se encuentra a escala con longitudes de rama en las mismas unidades de las distancias evolutivas utilizadas para inferir el árbol filogenético. Las distancias evolutivas se calcularon utilizando el método de probabilidad máxima compuesta (Tamura et al. 2004) y se encuentran en las unidades del número de sustituciones de bases por sitio. Se observó un total de 1,824,218 posiciones en el conjunto de datos finales. Los análisis evolutivos se realizaron con MEGA7 versión 7.0 (Kumar et al. 2016).

Figura 6 Árbol filogenético de máxima verosimilitud (MLE) bajo el modelo Tamura-Nei (Tamura & Nei 1993). Se muestra el árbol con la probabilidad de registro más alta (-24,308,138.44) El árbol se generó a través de la búsqueda heurística obtenida automáticamente aplicando los algoritmos de unión-vecindad y BioNJ a una matriz de distancias en pares estimadas, utilizando el método de máxima probabilidad de composición (MCL) y luego seleccionando la topología con un valor de probabilidad de registro superior. El árbol se dibuja a escala con longitudes de rama medidas en el número de sustituciones por sitio. Se observó un total de 1,824,218 posiciones en el conjunto de datos finales. Los análisis evolutivos se realizaron con MEGA7 versión 7.0 (Kumar et al. 2016).
Este resultado podría deberse al origen geográfico de cada individuo y su consiguiente evolución genética a través de los años (Motamayor et al. 2002). No obstante, si prestamos atención al resultado del análisis de máxima verosimilitud (Figura 6), la muestra denominada GAI01 se considera la rama primaria (out group) que da origen a los clados antes mencionados. De ser correcto lo anterior, la muestra GAI01 se ubicaría dentro de la población como el individuo con mayor rastro evolutivo pues las secuencias nucleotídicas de sus regiones conservadas son mayormente distantes en comparación con muestras que forman inclusive clústeres conjuntos (e.g. JGH02 y CCC03; MTJ01 y ALC03; MTJ03 y EJP01; JGH01 y MAJ01).
Tomando en cuenta dichos resultados la muestra GAI01 se encuentra directamente emparentada con JGH01 y MAJ01, aunque como se ha mencionado en repetidas ocasiones, las predicciones al ser heurísticas sólo sugieren una aproximación a la variabilidad genética real de las especies, por lo que el entendimiento de la filogenia y parentesco generacional entre individuos de cada clado podría cambiar a la luz de nuevas técnicas de secuenciación y estudios taxonómicos complementarios.
Análisis BLAST contra el genoma de referencia del cacao, regiones codificantes y predicción ontológica. Mediante la herramienta BLASTn se alinearon las secuencias de cada uno de los genomas reducidos contra el genoma de referencia del cacao, cuyos resultados bioinformáticos sugieren diversos porcentajes de homologías (% de identidad > 93) respecto a los cromosomas 1, 3, 5, 7, 8 y 9 del genoma de referencia (Tabla 2). De esta forma, las alineaciones de las secuencias de las muestras de cacao respecto a regiones codificantes referentes sugieren la presencia de homologías que van desde el a.c. 95 al 100 % con una longitud promedio > 150 pb (Tabla 3). Comparando datos del repositorio UniProtKB, se realizó una predicción heurística de la ontología de los péptidos traducidos a partir de las CDs del genoma del cacao Criollo (i.e. locus de referencia).
Tabla 2 Resultado del análisis BLAST contra el genoma de referencia del cacao.
| Muestra | Ref | % identidad | Longitud de coincidencia* | Posición inicial de ensamblado* | Posición final de ensamblado* | Posición inicial de referencia* | Posición final de referencia* |
|---|---|---|---|---|---|---|---|
| MTJ01 | chr1 | 99.56 | 226 | 106,467 | 106,692 | 23,570,132 | 23,569,908 |
| ACP03 | chr8 | 99.49 | 195 | 861,199 | 861,393 | 6,718,366 | 6,718,172 |
| JGH02 | chr9 | 93.99 | 233 | 316,953 | 317,184 | 25,425,291 | 25,425,059 |
| MTJ03 | chr1 | 100 | 225 | 106,465 | 106,689 | 23,570,132 | 2,356,9908 |
| JGH01 | chr1 | 100 | 225 | 106,635 | 106,859 | 23,570,132 | 23,569,908 |
| EJP01 | chr1 | 99.56 | 226 | 106,362 | 106,586 | 23,570,133 | 23,569,908 |
| LlHC02 | chr1 | 96.72 | 183 | 34,751 | 34,933 | 21,908,868 | 21,908,689 |
| ACP01 | chr7 | 99.47 | 187 | 1,141,078 | 1,141,264 | 11,836,804 | 11,836,618 |
| MAJ02 | chr1 | 99.56 | 226 | 106,556 | 106,781 | 23,570,132 | 23,569,908 |
| VAP02 | chr1 | 99.56 | 226 | 106,503 | 106,727 | 23,570,133 | 23,569,908 |
| CCC03 | chr1 | 99.56 | 226 | 106,465 | 106,690 | 23,570,132 | 23,569,908 |
| ALC03 | chr1 | 98.51 | 202 | 293,460 | 293,661 | 18,508,996 | 18,509,197 |
| EJP03 | chr1 | 99.56 | 226 | 106,641 | 106,866 | 23,570,132 | 23,569,908 |
| ALC02 | chr5 | 100 | 180 | 657,613 | 657,792 | 17,691,733 | 17,691,554 |
| MAJ01 | chr1 | 99.56 | 226 | 106,449 | 106,673 | 23,570,133 | 23,569,908 |
| VAP03 | chr7 | 99.53 | 213 | 476,016 | 476,228 | 15,981,285 | 15,981,497 |
| GAI02 | chr1 | 99.56 | 226 | 106,469 | 106,694 | 23,570,132 | 23,569,908 |
| MAJ03 | chr1 | 99.47 | 187 | 920,342 | 920,528 | 1,495,836 | 1,496,022 |
| GAI01 | chr3 | 100 | 206 | 441,310 | 441,515 | 25,969,724 | 25,969,519 |
| LlHC03 | chr1 | 98.52 | 203 | 293,284 | 293,486 | 18,508,995 | 18,509,197 |
* Posiciones expresadas en pares de bases (Pb)
Tabla 3 Resultado del análisis BLAST contra regiones codificantes del genoma de referencia del cacao.
| Muestra | Locus Ref | % de identidad | Longitud de coincidencia* | Posición inicial en ensamblado* | Posición final del ensamblado* | Posición inicial en la referencia* | Posición final en la referencia* |
|---|---|---|---|---|---|---|---|
| MTJ01 | c05v2_t023880.2 | 100 | 151 | 560,446 | 560,596 | 581 | 431 |
| ACP03 | Tc04v2_t013850.5 | 99.37 | 158 | 765,330 | 765,487 | 2,306 | 2,463 |
| JGH02 | Tc01v2_t002890.4 | 98.93 | 187 | 919,681 | 919,867 | 547 | 361 |
| MTJ03 | Tc05v2_t023880.2 | 100 | 151 | 560,498 | 560,648 | 581 | 431 |
| JGH01 | Tc03v2_t016000.1 | 100 | 151 | 832,866 | 833,016 | 672 | 522 |
| EJP01 | Tc09v2_t010110.1 | 95.65 | 230 | 573,644 | 573,873 | 645 | 416 |
| LlHC02 | Tc03v2_t016000.1 | 100 | 151 | 832,520 | 832,670 | 672 | 522 |
| ACP01 | Tc10v2_t006710.1 | 100 | 151 | 1,091,751 | 1,091,901 | 801 | 651 |
| MAJ02 | Tc04v2_t013850.1 | 100 | 158 | 764,981 | 765,138 | 2,306 | 2,463 |
| VAP02 | Tc01v2_t002890.4 | 98.93 | 187 | 919,752 | 919,938 | 547 | 361 |
| CCC03 | Tc05v2_t023880.2 | 100 | 152 | 560,952 | 561,103 | 581 | 430 |
| ALC03 | Tc05v2_t023880.2 | 100 | 152 | 560,866 | 561,017 | 581 | 430 |
| EJP03 | Tc03v2_t016000.1 | 100 | 151 | 832,880 | 833,030 | 672 | 522 |
| ALC02 | Tc02v2_t023880.2 | 100 | 151 | 560,668 | 560,818 | 581 | 431 |
| MAJ01 | Tc03v2_t026030.3 | 96.84 | 190 | 1,629,165 | 1,629,353 | 1,368 | 1,179 |
| VAP03 | Tc03v2_t026030.3 | 98.95 | 190 | 16,29,707 | 1,629,895 | 1,368 | 1,179 |
| GAI02 | Tc01v2_t014540.1 | 99.37 | 158 | 632,948 | 633,105 | 1,058 | 1,215 |
| MAJ03 | Tc01v2_t002890.4 | 99.47 | 187 | 920,342 | 920,528 | 547 | 361 |
| GAI01 | Tc01v2_t002890.4 | 99.47 | 187 | 920,204 | 920,390 | 547 | 361 |
| LlHC03 | Tc04v2_t013850.5 | 100 | 158 | 765,097 | 765,254 | 2,306 | 2,463 |
* Posiciones expresadas en pares de bases (Pb)
Entre otros datos interesantes, los resultados sugieren ontologías relacionadas con crecimiento y desarrollo de tejido meristemático (A0A061EM97); dominios relacionados con la presencia de dedos de zinc (Q6AVI0); ubiquitinas ligasa tipo 5 (023225); factores de transcripción tipo WRKY (Q8S8P5); proteínas de resistencia contra enfermedades tipo RGA3 (Q7XA40); proteínas de enlace-ADN tipo WRKY (D7LVF5); proteínas tioesterasas palmitol-acil acarreadoras cloroplásticas (Q9SJE2); superfamilias SWIM dedos de zinc (M9H780). Igualmente se hicieron presentes dominios de dedos de zinc tipo CCCH (Q9FNZ1); proteínas de resistencia tipo RPP13 (Q9M667); dominios tipo mano EF de enlace-calcio (F4IJ44); pirofosfatasas tipo 3 inorgánicas (A0A1S2YF43) y superfamilias fosfatidilinositol 3 y 4-kinasas (Q22H25).
Todas las ontologías anteriores se encontrarían relacionadas principalmente con el metabolismo secundario de la planta de cacao. Es importante mencionar que debido a la aún escasa caracterización proteómica del género Theobroma, al realizarse la búsqueda de ontologías de los loci traducidos del genoma de referencia, se encontró una basta cantidad de predicciones hipotéticas que hacen alusión a proteínas que pudieran relacionarse con diversos factores de transcripción e isoformas proteicas (Tabla 4).
Tabla 4 Predicción ontológica de péptidos traducidos a partir de CDs del genoma de referencia de cacao Criollo.
| Muestra | Locus ref | Inicio lectura Pb | Final lectura Pb | Predicción funcional |
|---|---|---|---|---|
| MTJ01 | Tc05v2_t023880.2 | 37041313 | 37046873 | Proteína hipotética (TSO1 tipo CXC 2) |
| ACP03 | Tc04v2_t013850.5 | 24255768 | 24284184 | Proteína hipotética (dominio dedos de zinc tipo BED) |
| JGH02 | Tc01v2_t002890.4 | 1493275 | 1499411 | Proteína hipotética (dominio U-box tipo 5) |
| MTJ03 | Tc05v2_t023880.2 | 37041313 | 37046873 | Glutaredoxina-C11 |
| JGH01 | Tc03v2_t016000.1 | 29589326 | 29591723 | Hipotético (factor de transcripción tipo WRKY 30) |
| EJP01 | Tc09v2_t010110.1 | 6312880 | 6314805 | Proteína sin caracterizar |
| LlHC02 | Tc03v2_t016000.1 | 29589326 | 29591723 | Hipotético (factor de transcripción tipo WRKY 30) |
| ACP01 | Tc10v2_t006710.1 | n/d | n/d | n/d |
| MAJ02 | Tc04v2_t013850.1 | 24254378 | 24284184 | Proteína tirosina sulfotransferasa |
| VAP02 | Tc01v2_t002890.4 | 1493275 | 1499411 | Proteína hipotética (dominio U-box tipo 5) |
| CCC03 | Tc05v2_t023880.2 | 37041313 | 37046873 | Proteína hipotética (TSO1 tipo CXC 2) |
| ALC03 | Tc05v2_t023880.2 | 37041313 | 37046873 | Proteína hipotética (TSO1 tipo CXC 2) |
| EJP03 | Tc03v2_t016000.1 | 29589326 | 29591723 | Hipotético (factor de transcripción tipo WRKY 30) |
| ALC02 | Tc02v2_t023880.2 | 34546680 | 34549916 | Proteína sin caracterizar At3g61260 |
| MAJ01 | Tc03v2_t026030.3 | 35650254 | 35653834 | Superfamilia dedos de zinc SWIM |
| VAP03 | Tc03v2_t026030.3 | 35650254 | 35653834 | Superfamilia dedos de zinc SWIM |
| GAI02 | Tc01v2_t014540.1 | 10887971 | 10896117 | Proteína sin caracterizar isoforma tipo 1 |
| MAJ03 | Tc01v2_t002890.4 | 1493275 | 1499411 | Proteína hipotética (dominio U-box tipo 5) |
| GAI01 | Tc01v2_t002890.4 | 1493275 | 1499411 | Proteína hipotética (dominio U-box tipo 5) |
| LlHC03 | Tc04v2_t013850.5 | 24255768 | 24284184 | Proteína hipotética (dominio dedos de zinc tipo BED) |
La figura 7A muestra una alineación de residuos de aminoácidos traducidos de los loci de referencia anteriores, cuyas regiones conservadas se representan mayoritariamente por ácido glutámico, serina, arginina, histidina, aspartato y tirosina. En la Figura 7B se observa la alineación completa de los 20 loci de referencia del genoma del cacao Criollo en donde se correlacionan secuencias similares e idénticas de aminoácidos entre los 800 y 900 residuos. Finalmente, mediante un análisis de filogenia mínima evolutiva (Figura 8) se calculó la longitud de las ramas del árbol óptimo obteniendo un valor de 1,288.2 sustituciones por sitio, donde se observa la presencia de tres taxones principales representados por cuatro muestras y un cuarto taxón compuesto por dos subgrupos de cuatro muestras cada uno.

Figura 7 A. Alineación de secuencias de aminoácidos de regiones codificantes. B. Representación general de secuencias altamente conservadas (rojo) y menos conservados (azul). El análisis se realizó con COBALT (Papadopoulos & Agarwala 2007).
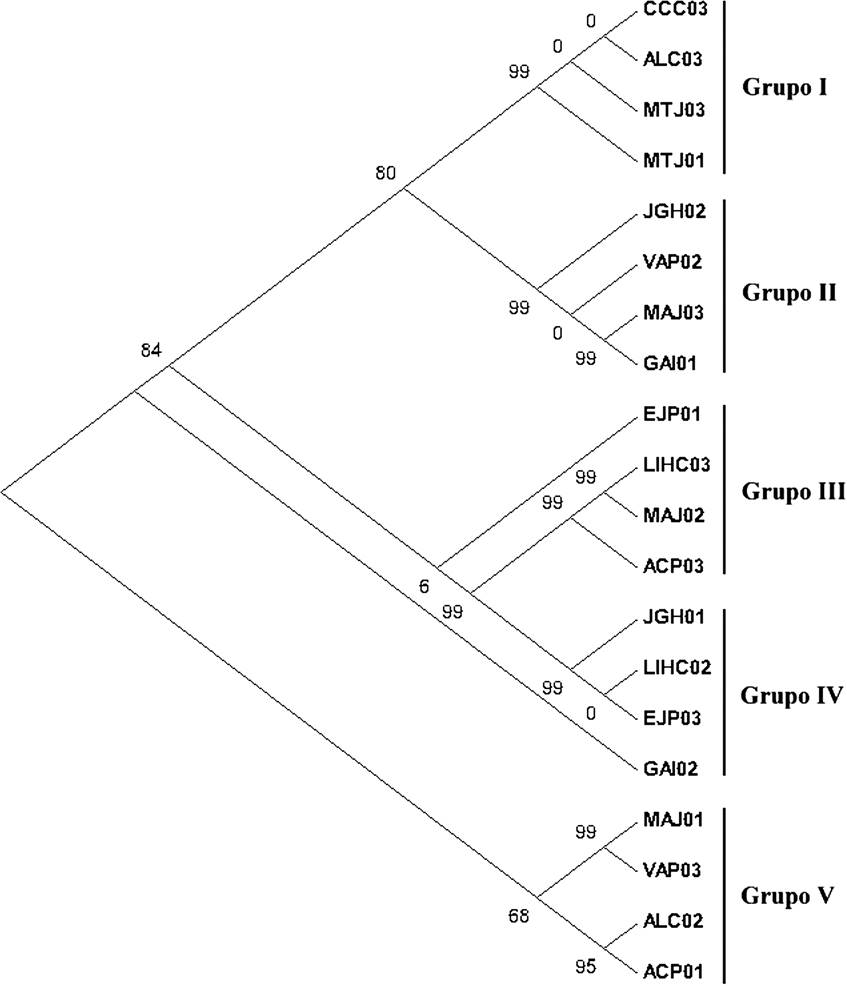
Figura 8 Diagrama de historia evolutiva mínima. El análisis se realizó utilizando el método Neighbor-Joining (Saitou & Nei 1987). Se muestra el árbol óptimo con una suma de la longitud de rama = 1,288.20604396. La probabilidad de confianza (X 100) de que la longitud de la rama interior es > 0, según la estimación mediante la prueba de arranque (se muestran 500 repeticiones junto a las ramas (Rzhetsky & Nei 1992). Las distancias evolutivas se calcularon utilizando el método del número de diferencias (Nei & Kumar 2000). Se observó un total de 321 posiciones en el conjunto de datos final. Los análisis evolutivos se realizaron en MEGA7 versión 7.0 (Kumar et al. 2016).
Discusión
La domesticación del cacao ha despertado el interés de un conjunto de diversas disciplinas. No obstante lo anterior, nuestro conocimiento sobre dicha tarea aún resulta incompleta, ya que a menudo ésta implica trabajar con pocos grupos genéticos, regiones geográficas sumamente específicas, información arqueológica fragmentada y, sobre todo en un sentido estrictamente molecular; con un número muy limitado de marcadores genéticos (Motamayor et al. 2002). La propuesta generalmente aceptada sobre el origen de la domesticación del cacao sugiere que éste se introdujo en Mesoamérica en la edad precolombina por la cultura olmeca, a partir de las variedades de cacao presentes en la alta Amazonía (norte de América del Sur), región considerada “el foco de diversidad” para el género Theobroma (Bartley 2005).
Otra propuesta interesante sugiere que la ruta de domesticación del árbol del chocolate se fragmentó a lo largo de la cuenca del Amazonas mediante dos vertientes: una en dirección norte y otra en dirección oeste (Motamayor et al. 2003). Según esta hipótesis, la domesticación del cacao se habría originado en América del Sur, extendiéndose a América Central y México a través de prácticas de comercio por parte de los nativos americanos (Stone et al. 1984). Cabe mencionar que la corriente antropológica apoya la propuesta de domesticación ocurrida en Mesoamérica (Powis et al. 2011). En este sentido, la cruza continua entre árboles de cacao domesticados y silvestres dio origen de manera reciente a los clústeres genéticos conocidos (Zhang et al. 2012). Por tal razón el impacto de los antiguos procesos de domesticación e hibridación moderna forman parte de la historia evolutiva del género Theobroma.
Para profundizar en el campo de la genómica de esta especie y en su variabilidad genética poblacional, se recurre de manera reciente a secuenciaciones de nueva generación, incluyendo también el estudio de cultivos de importancia económica a través del ensamblaje y secuenciación de novo de sus genomas. En este trabajo se recurrió a la explotación de algoritmos fundamentados en el diagnóstico de paralelismos masivos (i.e. secuenciación en paralelo de millones de transcritos), metodología equiparable a la mayoría de las actuales secuenciaciones de nueva generación.
Esta tecnología brinda mejores oportunidades para estudiar a fondo la genómica estructural y funcional de distintas especies, a través de la incursión en las disciplinas ómicas (e.g. transcriptómica, proteómica y metabolómica) (Elshire et al. 2011). Gracias al descubrimiento de marcadores moleculares generados por NGS y diversos análisis bioinformáticos, es posible explorar la diversidad genética y filogenia evolutiva de cientos de cultivos vegetales mediante la secuenciación parcial o completa de sus genomas.
El genotipado por secuenciación llevado a cabo en nuestro trabajo propone una aproximación a la secuenciación multiplexada de fragmentos de ADN escindidos mediante REs que contienen secuencias de nucleótidos cortas y únicas (códigos de barras) a través de un solo canal de secuenciación (Craig et al. 2008). Este enfoque conocido como cizallamiento aleatorio de ADN funciona muy bien para especies con genomas relativamente pequeños, incluido el ADN de organelos. En el caso de las variedades nativas de cacao provenientes de Tabasco y Chiapas fue posible explorar regiones sumamente pequeñas y conservadas. Dicho enfoque se ha utilizado para determinar rápidamente secuencias completas del genoma de cloroplastos de abetos, así como varias especies del género Pinus (Cronn et al. 2008) y también, para la caracterización y mapeo de SNPs en arroz (Huang et al. 2009).
Hoy en día, distintas herramientas biotecnológicas de alto rendimiento y bajo costo suelen ser accesibles para la mayoría de los investigadores. Debido a lo anterior, diversas disciplinas genómicas se encuentran a menudo relacionadas con otras ramas de las ciencias naturales y sociales, incluyendo por ejemplo, la soberanía y seguridad alimentaria, entre otras (Ricaño-Rodríguez et al. 2018, Godfray et al. 2010). Por mencionar un ejemplo importante, la caracterización molecular de distintas variedades de cacao ha permitido descubrir polimorfismos de un solo nucleótido en su genoma, cuyo objetivo es la determinación heurística de la huella evolutiva de la especie, ya que los SNPs (aunque muy escasos) son la clase más conservada de secuencias de ADN en los genomas de las plantas (Buckler & Thornsberry 2002).
En un ensayo de caracterización genética, la correcta selección de enzimas de restricción que serán utilizadas para la obtención de transcritos mediante PCR, resulta determinante para la obtención de resultados favorables. En dicho sentido, las enzimas Bgl II y Ddel I poseen ventajas destacables, por ejemplo: son aplicadas bajo metodologías fáciles, rápidas y baratas, además de que son extremadamente específicas en los extremos de corte del genoma, pero lo más importante es que permiten el estudio de regiones sumamente cortas y difíciles de alcanzar en comparación con otros métodos comunes de diagnóstico molecular, como por ejemplo los marcadores microsatelitales o espaciadores internos transcritos que suelen abarcar secuencias genéticas conservadas mucho mayores (Buckler & Thornsberry 2002). Así, la elección adecuada de enzimas restrictivas para cada generación de bancos de genes, disminuye las posibilidades de que se creen fragmentos de nucleótidos con secuencias repetitivas durante el proceso de termociclación (Gore et al. 2007, 2009), lo cual, en la etapa de análisis bioinformático, reduce enormemente los problemas de alineación entre secuencias sujetas a análisis filogenéticos.
En comparación con otros tipos de marcadores moleculares (e.g. ISSR; Inter Simple Sequence Repeats, RFLP; Restriction Fragment Length Polymorphism, AFLP; Amplified Fragment Length Polymorphism, ITS; Internal Transcribed Spacer), los ensayos con SNPs se realizan sin separar el ADN por tamaño y, por lo tanto, se automatizan en formatos de placas de ensayo o microchips. Generalmente la naturaleza bialélica de los SNPs (Livingstone et al. 2012, Dadzie et al. 2013, Ji et al. 2013) resulta en una tasa de error mucho menor y la genotipificación se puede multiplexar, lo que permite secuenciaciones más rápidas a un costo menor.
En los últimos años, aunque de manera muy limitada, se han desarrollado marcadores de SNPs para potenciar el mejoramiento de la producción de cacao y el respectivo manejo de germoplasma a través del estudio de su variabilidad genética (Allegre et al. 2012, Kuhn et al. 2012, Livingstone et al. 2012, Takrama et al. 2012). Sin embargo, los antecedentes sobre este tipo de estudios en el género Theobroma aún son relativamente escasos. Por mencionar algunos ejemplos importantes, Ji et al. (2013) identificaron un conjunto de SNPs derivados de la caracterización de marcadores de secuencias expresadas (EST; Expressed Tag Sequences) en cacao, cuyo resultado identificó variedades nicaragüenses y hondureños con mayor variabilidad genética. Asimismo, de manera reciente Lindo et al. (2018) realizaron la caracterización molecular de germoplasma de cacao proveniente de Jamaica utilizando marcadores SNPs.
De acuerdo con lo antes mencionado, los resultados de variabilidad alélica polimórfica de las muestras estudiadas en este proyecto abren la posibilidad de seleccionar especímenes con mayor presencia de SNPs para ser reproducidas mediante injertos en invernaderos, o bien, para cultivarse como bancos de germoplasma con fines de conservación in situ o ex situ. Lo anterior motivaría el desarrollo de sistemas diversificados mayormente pertinentes a las condiciones del trópico (Hipólito-Romero et al. 2017). Es bien sabido que el éxito de perpetuidad de una especie depende de su capacidad de adaptación para con los factores bióticos y abióticos interactuantes, y en dicho sentido, diversos estudios transcriptómicos del género Theobroma arrojan teorías bastante interesantes respecto a la correlación entre su variabilidad genética y su capacidad de supervivencia.
Entre los primeros intentos de secuenciación genómica del cacao, se encuentra aquél realizado en la variedad Matina, anotada hasta ahora casi en su totalidad y cuyo tamaño oscila alrededor de los 445 Mpb (el genoma de la variedad criolla posee 430 Mpb) (Motamayor et al. 2013). Gracias a estos antecedentes se han caracterizado distintos genes de interés biotecnológico y agrícola (Ricaño-Rodríguez et al. 2018), lo que beneficia principalmente al campo de la fitopatología, pues el correcto estudio de su estructura nucleotídica dilucida la naturaleza de sus respuestas epigenéticas desencadenadas frente a la interacción con organismos antagónicos. Respecto a la interacción a nivel metabólico entre los géneros Theobroma y Moniliophthora, vale la pena mencionar que la severidad de la infección por parte del hongo fitopatógeno sobre la planta, depende en gran medida de los efectores de virulencia que sean expresados por Moniliophthora, los cuales a su vez, se verán afectados por la cantidad de polimorfismos que se encuentren presentes en el genoma de Theobroma y que activarán en consecuencia su mecanismo de defensa a nivel molecular (Jones & Dangl 2006).
La interacción entre Moniliophthora y Theobroma desencadena la expresión (de ambos genomas) de distintos alelos que codifican principalmente péptidos involucrados en rutas metabólicas de respuestas sistémicas. Por parte del hongo, algunas de éstas se relacionan con procesos de fitopatogenicidad al igual que transposición replicativa y transcripción (e.g. citoquininas deshidrogenasas, glicósido hidrolasas, proteínas I3, proteínas ricas en cisteína, peptidasas, transposasas y thaumatina) (Ricaño-Rodríguez et al. 2018). Por otra parte, el umbral de resistencia de la planta de cacao que resulta infectada dependerá en gran medida de la presencia de regiones polimórficas en su genoma, pues a mayor variabilidad genética, mayor posibilidad de transcribir efectores moleculares involucrados en su mecanismo de defensa (Jones & Dangl 2006).
En los últimos años, muchos productores alrededor del mundo se valen de herramientas biotecnológicas para identificar genes de resistencia en el cacao que combatan enfermedades fitopatológicas, como por ejemplo: la coloquialmente conocida “escoba de bruja” (WBD; Witche´s Broom Desease) causada por Moniliophthora perniciosa. Así, el mejoramiento de variedades vegetales resistentes a plagas es deseable para generar bancos de genes involucrados en mecanismos de defensa, a fin de reducir las posibilidades de que el fitopatógeno desequilibre el metabolismo sistémico de su hospedador. Por esta razón, obtener nuevos marcadores moleculares para seleccionar genotipos de interés de cacao es una estrategia potencial para acelerar los programas de propagación de la especie (Jones et al. 2002).
En dicho sentido, Lima et al. (2009) identificaron regiones codificantes en el genoma de Theobroma mientras estudiaban la presencia de SNPs haciendo uso de marcadores moleculares EST. Además de este tipo de estudios se ha identificado el fenómeno de polimorfismo no neutral, el cual se produce en la secuencia de codificación o en sus proximidades y está directamente relacionado con la variabilidad genética (Gesteira et al. 2007).
Como ya se mencionó, la secuenciación del genoma del cacao dio como resultado el descubrimiento de cientos de marcadores SNPs (Argout et al. 2017). Esto permitirá a futuro generar mapas genéticos de la especie más específicos con una mayor precisión de loci de carácter cuantitativo (QTL; Quantitative Trail Locus). Los análisis BLAST de las secuencias de muestras provenientes de distintas regiones cacaoteras de los Estados de Tabasco y Chiapas (Figura 1) mostraron altos porcentajes de homologías respecto a su alineación con regiones codificantes conservadas y sus respectivos transcritos cuando éstas se compararon con el genoma de referencia del cacao (Tablas 2 y 3).
Entre los resultados más interesantes sobre predicción funcional de los genomas de las variedades de cacao estudiadas en este proyecto, se observó la presencia de dominios de dedos de zinc cuya función podría relacionarse con motivos proteínicos que coordinan iones que estabilizan sus pliegues y, dado que estos funcionan como módulos de interacción entre ADN y ARN (Jones & Dangl 2006), su predicción se relacionaría con supuestos factores de transcripción de la planta.
Así también, la predicción funcional heurística de los transcritos obtenidos mediante la generación de bancos de genes sugiere ontologías relacionadas con crecimiento y desarrollo de tejido meristemático, ubiquitinas ligasa tipo 5 que actúan como reguladores sistémicos en la interacción planta-fitopatógeno, resistencia contra enfermedades tipo RGA3 y tioesterasas palmitol-acil acarreadoras de cloroplastos, entre otras. Por otra parte, se observó una distribución funcional que muestra dominios de dedos de zinc tipo CCCH con un posible papel estructural (Q9FNZ1); proteínas de resistencia tipo RPP13 (Q9M667); dominios tipo mano EF de enlace-calcio (F4IJ44); pirofosfatasas tipo 3 inorgánicas que catalizan la ruptura de enlaces altamente energéticos entre grupos fosfatos (A0A1S2YF43) y superfamilias fosfatidilinositol 3 y 4-kinasas que suelen transportar ATP y grupos fosforilos (Q22H25) (Gesteira et al. 2007).
Dado que los estudios moleculares del cacao complementados con técnicas de GBS son escasos, muchas regiones genómicas codificantes de variedades mexicanas no se encuentran aún caracterizadas, por lo que su secuencia respectiva no coincide con datos previamente depositados en el repositorio del NCBI, razón por la cual la predicción ontológica resulta en ocasiones hipotética. Lo anterior brinda la oportunidad de realizar futuras caracterizaciones y anotaciones funcionales de nuevas regiones codificantes conservadas del genoma del género Theobroma. En este trabajo, mediante la técnica de GBS se generaron biobancos que fueron caracterizados a través de distintos análisis bioinformáticos, y fue posible estudiar a fondo un fragmento de la historia evolutiva de la especie en cuestión incluyendo la caracterización de algunas de sus regiones génicas codificantes conservadas, lo cual es un factor determinante en la expresión transcripcional del género Theobroma (Rafalski 2002).
Los ensayos enfocados en la detección de SNPs en cacao mediante el método GBS identifican haplotipos útiles en estudios de variabilidad genética alrededor del mundo. En resumen, dichos resultados se traducen en la obtención de balances de rendimiento de producción, simplicidad, ahorro de costos y también; transferencia de datos moleculares de vital uso tanto para los productores como genetistas poblacionales.
De alguna manera, los análisis de parentesco molecular y filogenia evolutiva de plantas han permitido a los productores de germoplasma y fitomejoradores, la generación de cultivos perennes con patrones de polinización y flujo de genes altamente rentables (Ashley 2010, Lacombe et al. 2013, Takrama et al. 2014), lo que lleva a comprender mejor los procesos de domesticación y selección de individuos resilientes (Motamayor et al. 2002). En el caso del cacao, ciertas variedades de Trinidad y Tobago (que forman parte del germoplasma mejorado de cacao en África occidental) han mostrado altos registros de reproducción lo que se correlaciona en gran parte con la presencia de SNPs (Takrama et al. 2014).
Mediante análisis genéticos poblacionales que utilizan marcadores microsatelitales se ha descubierto un gran número de grupos genéticos, con una clara diferenciación entre variedades que se encuentran en la cuenca del Amazonas y variedades criollas situadas en América Central. Aunque no sabemos con total certeza cuál es el origen genético de las variedades genotipificadas en este estudio, mediante análisis de unión de secuencias nucleotídicas vecinas (NJ) (Figura 4), agrupamiento jerárquico aglomerado simple con media aritmética (UPGMA) (Figura 5) y máxima verosimilitud (MLE) (Figura 6), determinamos que más del 50 % de árboles replicados en los taxones agrupados en la prueba de arranque fue muy similar entre si, lo que indica una jerarquización equilibrada entre las muestras con una distribución uniforme discreta (Tamura & Nei 1993, Kumar et al. 2016).
Aunado a lo anterior, el estudio de evolución mínima (Figura 8) (Rzhetsky & Nei 1992) muestra un reagrupamiento categórico con árboles replicados casi al 100 %, por lo que creemos que la población pasó por un “cuello de botella” en algún momento de su historia evolutiva, debido muy probablemente a las cruzas constantes entre individuos circundantes, así como a las condiciones ambientales desfavorables que forzaron el aumento de la variabilidad alélica de la población como un mecanismo de supervivencia (Motamayor et al. 2008).
Teniendo presentes los antecedentes sobre el origen genético del cacao aún existe una gran brecha respecto al conocimiento de su proceso evolutivo, lo que dificulta proponer escenarios claros para la domesticación de la especie. Aunque es ampliamente aceptada la teoría de que las poblaciones ecocéntricas mesoamericanas contribuyeron en los últimos tiempos a la composición genética actual de Theobroma, la mayoría de los cultivares de cacao siguen siendo endémicos. En principio, la especie más estrechamente relacionada con T. cacao en términos genéticos es Theobroma grandiflorum (Richardson et al. 2015), aunque curiosamente las características morfológicas de los árboles y los frutos de esta última especie son muy diferentes a las encontradas en T. cacao (Carvalho-Santos et al. 2012).
El aumento de la frecuencia de alelos deseables en una población vegetativa, es un requisito clave para lograr ganancias reproductivas a largo plazo. En este sentido, las huellas moleculares de las colecciones de germoplasma de cacao en el mundo han mejorado el conocimiento de las relaciones genéticas entre accesiones, que sirven para guiar la selección de líneas mejoradas reduciendo las posibilidades de introgresión de alelos indeseables (Collard & Mackill 2008). Por consiguiente, la aplicación de marcadores SNPs en combinación con técnicas de genómica funcional y fenotipificación han acelerado el ritmo y ganancias de la propagación de cultivos arbóreos, además de mantener la resiliencia y desarrollo sostenible de especies comerciales.
Por último, no omitimos mencionar que éste es el primer genotipado por secuenciación de variedades de cacao nativo mexicano que se realiza en nuestro país, cuyo BioProyecto se encuentra registrado ante el Centro Nacional para la Información Biotecnológica (accession number: SAMN11334316).

