











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
Disorders of sexual development (DSD) were defined in the Chicago Consensus Meeting, 2005, and published as the Chicago Consensus Statement, 2006, as “congenital conditions in which the development of chromosomal, gonadal, and anatomical sex is atypical".1
Mutations in regulatory genes controlling urogenital ridge development, bipotential gonad formation, and cell differentiation in testicular or ovarian tissue have been identified among molecular causes for DSD. Several genes involved in the testis differentiation pathway have been described, along with their interactions. Knowledge of the ovary differentiation pathway, however, is limited.2,3
Diagnosis of DSD is difficult due to the great variability of clinical manifestations, late onset of some symptoms, and apparent discordancy in laboratory findings. Despite advancements in gene identification, there is still limited knowledge of the number of genes involved in DSD. This creates difficulties in the etiology workup of these conditions. Physicians are unable to establish a definitive diagnosis for many individuals.2,4-6
Significant advances in the understanding of molecular causes for DSD called for integration of new knowledge in the classification of these conditions. The most recent classification was implemented in the Chicago Consensus Conference, 2005. It sets up three principal groups: 1) DSD with sex chromosome abnormality, which includes Turner’s syndrome (45,XO), Klinefelter syndrome (47,XXY), Mixed gonadal dysgenesis, and chimeras. 2) DSD with 46,XY karyotype and 3) DSD with 46,XX karyotype without numeric or structural abnormalities.1
Limited knowledge of DSD pathophysiology and etiology, especially regarding the ovarian differentiation pathway, drives the need for thorough analysis of individuals with 46,XX karyotype and suspected DSD. A comprehensive study would diagnosis and proper management in the majority of these cases. This paper provides an updated of concepts about DSD with 46,XX karyotype, supplementing current knowledge of this condition.
Sexual determination and sexual differentiation
Sexual differentiation during prenatal development involves processes triggered and regulated by a large number of genes, proteins, and hormones. Phenotypical sex in humans depends on gonad development in the embryo. This process is primarily determined by the genetic constitution of the individual.2
Sexual development is divided into two processes: Sexual determination and sexual differentiation. In sexual determination, certain genes lead development of the undifferentiated bipotential toward ovary or testis. Sexual differentiation is mediated by hormones produced by the gonads during their formation and after completion (Figure 1).7
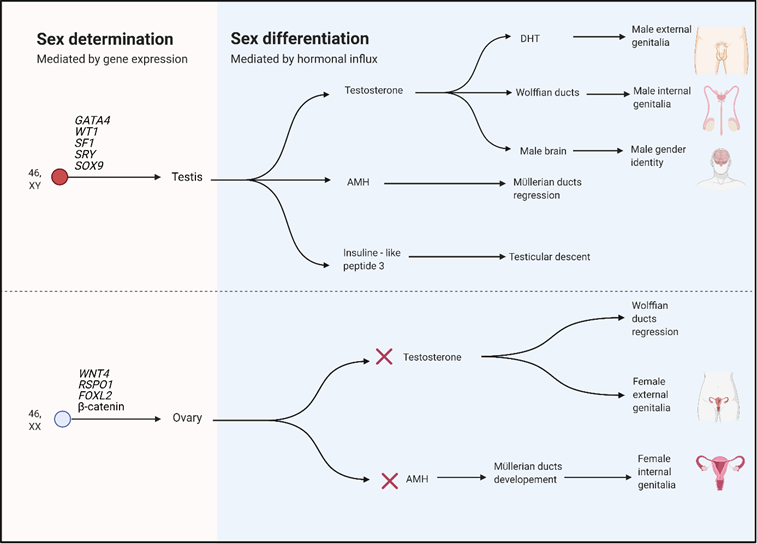
Adapted from Kremen et al.3 Created with BioRender.com
Figure 1 Sex determination and differentiation in humans
Many genes involved in sexual determination are also responsible for gonadal steroidogenesis affecting sexual differentiation. Any genetic or environmental influence disrupting the normal course of these processes may result in a DSD.4
During embryogenesis, the urogenital system derives from the urogenital ridge in the intermediate mesoderm around week 4 of gestation. The undifferentiated gonad arises from the mesonephric ventromedial surface from the intermediate mesoderm. The primordial germ cells migrate from the extraembryonic ectoderm (epiblast) along the dorsal mesentery of the posterior intestine. They reach the gonadal ridge toward the end of week 5, when they begin aggregation and proliferation (shown in Figure 2).2
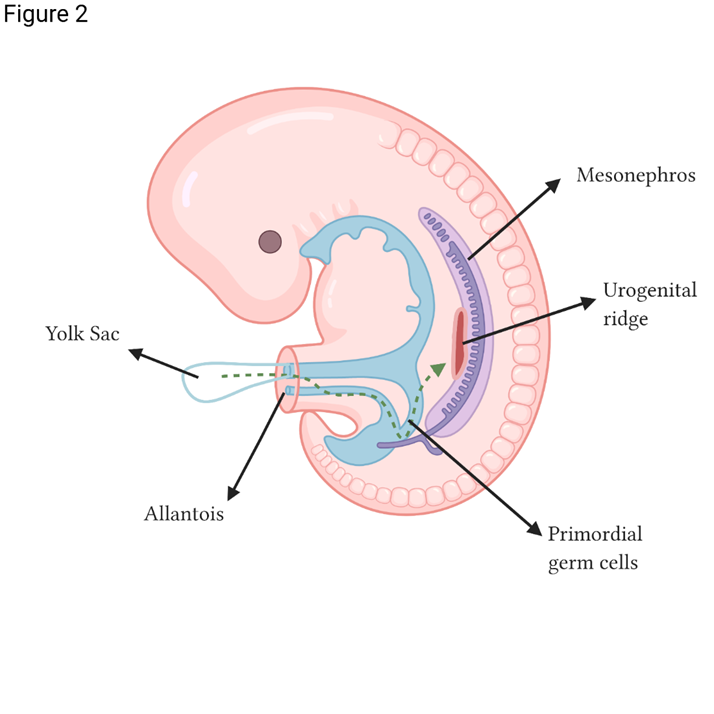
Adapted from Kousta et al.2 Created with BioRender.com
Figure 2 Primordial germ cell migration from the yolk sac to the urogenital ridge
During the sexual determination stage, the undifferentiated bipotential gonad starts responding to the stimulus produced by the increased expression in the gonadal ridges of several genes, including WT1, DAX1, SF- 1, LHX9, LIM1, PAX2, GATA4, EMX2, and WNT4. Those genes contribute at the onset of gonadal differentiation, whether the testis-specific pathway is activated or repressed. (A repressed testis pathway results in ovarian development).2,4
Several genes play important roles in development of the bipotential gonad, notably the nuclear receptor steroidogenic factor 1, SF-1 (also known as NR5A1) and the Wilms tumor suppressor (WT1) [5]. The SF-1 is relevant for the synthesis of adrenal and gonadal steroids (cytochrome P-450 steroid hydroxylase and 3β-hydroxysteroid dehydrogenase). The SF-1 is expressed in the bipotential gonad, and expression continues in the testis but is repressed in the ovary. It is also expressed in the developing urogenital ridge, the hypothalamus, the anterior pituitary gland, and the adrenal glands. Mutations in SF1 in 46,XY individuals may result in adrenal and variable degrees of gonadal insufficiency; in 46,XX individuals, they may result in adrenal insufficiency and apparently normal ovarian differentiation.8
The WT1 is a transcription factor necessary for the secondary development of the bipotential gonad. The WT1 action is important for mesenchymal-epithelial interactions in the Sertoli cells and Leydig cells. The WT1 is expressed in the urogenital ridge, mesonephros, kidney, gonad, and in the granulosa cells in females. Mutations of WT1 in 46,XY patients are associated with gonadal digenesis and kidney abnormalities (Frasier syndrome, Deny-Drash syndrome, and WAGR syndrome); in 46,XX individuals, those mutations do not have any effect on the gonadal development, but they may cause kidney abnormalities, such as focal and segmental glomerulosclerosis, and predispose individuals to Wilms tumor.2
Other genes involved in the development of the bipotential gonad are DMRT1, PAX2, and LHX9, which relate to the proliferation of somatic cells in the urogenital ridges. Pathogenic variants have been associated with disorders in somatic cell proliferation in the urogenital ridge, preventing gonad formation. Other genes, such as SOX3, SOX9, FGF9, and PGD2, promote the testis pathway. Alternately, the genes DAX1, WNT4, FOXL2, RSPO1, and CTNNB1 promote the ovarian differentiation pathway.2,5
Ovarian development
Despite efforts to fully understand ovarian development, the number of genes involved in the female pathway are still unknown, along with their functions and interactions.2
In the absence of SRY, female differentiation occurs with the female gonad germinal cells starting meiosis. Without SRY expression, SOX9 does not reach a critical expression threshold. This, combined with the expression of other genes such as RSPO1, WNT4, and FOXL2, leads differentiation towards ovary formation. In the female gonads, the WNT4 and RSPO1 transcription factors favor and stabilize the expression of the transcription factor CTNNB1 (B-catenine), which suppresses SOX9 expression and regulates transcription of WNT4-dependent genes. In this way, WNT4 expression antagonizes the male pathway.2,4
Research shows that deletions in the SOX9, SF-1, and WT1 genes, as well as duplications in DAX1 and WNT4 genes, may result in 46,XY gonadal dysgenesis.9-11 Conversely, in the absence of SRY, duplications of the SOX9 or SOX3 genes may lead to testicular 46,XX DSD.5,12
Disorders of sexual development
As mentioned, disruption of the processes of sex determination or sexual differentiation by any genetic or environmental factor may result in a DSD with discordance among chromosomal, gonadal, and anatomical sex. Such discordance will have different expressions in the phenotype, including abnormalities in the internal genitalia (absence or abnormal characteristics) and/or external genitalia (insufficient or excessive virilization).1,4,13
Those genitalia abnormalities may be evident at birth, with genital ambiguity or discordance between genotypic and phenotypic sex. Possible further manifestations are delayed puberty, amenorrhea, or insufficient/excessive virilization. Later in life symptoms may be infertility or premature menopause. Besides symptoms related to sexual development, abnormalities may exist in other systems, some as critical as adrenal insufficiency, which is potentially lethal without timely and precise management.4
Gender assignation in newborns is an important consideration. Opportune approach by a multidisciplinary team is fundamental to help define this aspect.6
Disorders of sexual development in 46,XX individuals
According to the Chicago consensus classification summarized in Table 1, the following DSD with 46,XX karyotype can be found:
Table 1 Classification of DSD in the 46,XX individual
| DSD in the 46,XX individual | ||
|---|---|---|
| 1. Disorders of gonadal (ovarian) development | 2. Androgenic excess | 3. Other abnormalities |
| 1.1 Ovotesticular DSD | 2.1. Fetal | 3.1 Mayer-Rokitansky-Küster-Hauser syndrome |
| 1.2 Testicular DSD | 2.1.1 Congenital adrenal hyperplasia | 3.2 Cloacal exstrophy, vaginal atresia, MURCS, other syndromes |
| 1.3. Gonadal dysgenesis | a. 21-hydroxylase deficit | |
| b. 11-B hydroxylase deficit | ||
| c. 3β-hydroxysteroid dehydrogenase deficit | ||
| d. P450 oxidoreductase (POR) deficiency | ||
| 2.2 Fetoplacental | ||
| 2.2.1 Aromatase deficiency | ||
| 2.3. Maternal | ||
| Luteoma, androgen-producing tumors |
1. Disorders of gonadal (ovarian) development
1.1 Ovotesticular DSD
This disorder is characterized by the presence of ovarian and testicular tissue in the same individual. It is possible to find normal ovarian tissue with numerous follicles and to find normal testicular tissue with seminiferous tubules containing germinal cells. As the individual develops, the testicular tissue tends to regress, which results in dysgenesis, interstitial fibrosis, and absence of spermatogonia. The presence of both tissues in the same gonad is called ovotestis. It is also possible to find ovaries and testes separated in the same individual, most frequently the ovarian tissue in the left side and testis tissue in the right side.2,14-16
Some patients exhibit a mix of derivatives from the Müller and Wolffian duct. In such cases, reports indicate that external genitalia are usually ambiguous with various degrees of virilization. Normal female genitalia may also be present.2
The SRY gene has been detected in one-third of patients with ovotesticular DSD. The presence of SRY would result from translocation of the gene to one of the X chromosomes during the paternal meiosis. The specific cause for this anomaly is still under study. The SRY gene may also be absent in a percentage of individuals, making it possible that other mechanisms are involved in creating the presence of two types of gonadal tissue in the same individual. These mechanisms remain unknown, but they may include germinal mosaicisms with mutations containing Y sequences confined to the testicular tissue. Also, the early presence of SRY may be enough for testis differentiation, even though its expression may disappear later in development.14
1.2 46,XX testicular DSD
Also referred to as De La Chapelle syndrome, this rare condition affects 1:20,000-1:25,000 male births. The phenotypical characteristic is normal male external genitalia in a majority of patients, but with up to 15% of individuals presenting ambiguous genitalia. Some present hypospadias in various degrees. After puberty, 85% of patients develop normal pubic hair, and penis size may also be normal, although with hypergonadotrophic hypogonadism. Some patients may also present gynecomastia and infertility (azoospermia). A small percentage exhibit chriptorchidia and/or anterior hypospadias. Gender identity is male. Regarding etiology, 90% of cases result from translocation of the Y chromosome material, including the SRY gene, to the X chromosome.17-21 Individuals with phenotypically positive SRY seldom present genitalia abnormalities. Also, these patients are reported with gynecomastia less often than SRY negative 46,XX patients.22
The SRY is absent in 10% of individuals with De La Chapelle syndrome, and the etiology in such cases remains unclear. Studies have identified, in the absence of SRY, duplications in the SOX9 gene leading to its overexpression and consequent differentiation of the testis pathways.12,20,23-28 Partial 22q duplications (duplications of bands from 22q11.2-22q13), overexpression of the SOX10 gene in 22q13, and mutations in the gene RSP01 have been associated with 46,XX testicular DSD, along with palmoplantar hyperkeratosis and predisposition to squamous cell skin carcinoma.29
The literature reports cases of individuals in the same family with 46,XX ovotesticular DSD and 46,XX testicular DSD. These findings indicate the possibility that these disorders are products of the same genetic error but with variability in phenotypic expression. Also, yet unknown environmental factors, known as endocrine disruptors, may modify the interaction among genes.30
1.3 Gonadal dysgenesis
In this type of disorder, the phenotype is female, but patients usually do not reach puberty and do not develop secondary sexual characteristics. The presentation may be sporadic or familiar. In familiar cases, an autosomal recessive inheritance pattern has been observed. The initial cause for consultation in patients with gonadal dysgenesis may be primary amenorrhea, delayed puberty, or infertility. Gonadal development is variable. There can be mild changes, from streak gonads to more severe changes, such as severe ovarian hypoplasia with just a few oocytes. Some patients may present a hypergonadotropic hypogonadism with elevated plasma levels of FSH and LH.
There are multiple etiologies for gonadal dysgenesis. In two-thirds of cases, dysgenesis is due to a genetic cause. The rest may be due to infections, infarction, or autoimmune disease. In some familiar cases, gonadal dysgenesis or premature ovarian insufficiency has been reported in patients with mutations in several loci in Xq (including BMP15, FMR1, and QM) and in Xp11.2-p.22.1, all related to ovarian development.2,31 Mutations in the gene FSHR have also been associated with sporadic and familiar cases of gonadal dysgenesis.
Other genetic syndromes, such as Perrault syndrome, have been associated with gonadal dysgenesis. Patients with this syndrome present associated neurosensory hypoacusia, achondroplasia, and lethal lung fibrosis with immunodeficiency. These patients do not have a particular risk of gonadal neoplasia.32
2. Disorders related to androgenic excess, formerly named female pseudohermaphroditism
It is necessary to know the process of steroidogenesis to understand the phenotypical manifestations of this group of disorders.
Steroidogenesis is the process in which cholesterol transforms into biologically active steroid hormones. Steroid hormones are produced in the adrenal cortex (glucocorticoid, mineralocorticoids), testicles (androgens), ovaries (estrogens), and some peripheral tissues (fat tissue, brain).33,34
The adrenal cortex has 3 zones: 1) Zona glomerulosa, located towards the periphery, in which mineralocorticoids synthesis takes place 2) Zona fasciculata, responsible for glucocorticoid production 3) Zona reticularis, in charge of adrenal androgen production.35 There are 2 types of steroidogenic enzymes: The cytochrome P450 (CYP) family and the hydroxysteroid dehydrogenases (HSD). The enzymes of the CPY family comprise type 1 (located in the mytochondriae) and type 2 (located in the endoplasmic reticulum). The HSD are categorized in aldo-keto reductase and short-chain dehydrogenases/reductases.33 Some enzymes may participate in extra-adrenal steroidogenesis, which results in the variety of possible phenotypical manifestations in patients with disorders in these enzymes.1,33
The steroid production pathway in the adrenal gland involves several enzymatic steps. First, cholesterol transport from the outer to the inner mitochondrial membrane is mediated by the steroidogenic acute regulator (StAR) protein. Then the cleavage of the lateral chain of the cholesterol by the cholesterol cleavage enzyme (coded by the gene CYP11A1) mediates the first and rate-limiting conversion step from cholesterol to pregnenolone.36 Other enzymes and cofactors, depending on the type of steroid to be produced, mediate further steps.
Below are the principal DSD with 46,XX karyotype related to disorders of steroid enzymes.
2.1.1. Congenital adrenal hyperplasia
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder that comprises a group of diseases caused by a deficiency in the enzymes responsible for cortisol and aldosterone biosynthesis in the adrenal glands. The CAH was first described by De Crecchio in 1865 in the necropsy of a male with 46,XX karyotype who died of an adrenal crisis.37
Patients with CAH may exhibit different clinical manifestations depending on the underlying enzyme deficiency: 21-hydroxylase, 11β-hydroxylase, 3β-hydroxysteroid dehydrogenase, or 17α- hydroxylase. Mutations in genes encoding these enzymes cause a deficiency in glucocorticoid and mineralocorticoid production, with an increase in ACTH levels, resulting in an excess of adrenal androgens and adrenal hyperplasia.2
2.1.1.1 21-hydroxylase deficiency
Deficiency of 21-hydroxylase (CYP21A2) leads to an accumulation of 17-hydroxyprogesterone (17OHP) and a consequential increase of the Δ4 androstenedione (Δ4 Α). Excessive accumulation of 17OHP amplifies the androgen production pathways leading to masculinization of the female fetus. Depending on the degree of functional loss of the 21-hydroxylase, the CAH adopts either a classic or non-classic clinical form.5,36
The 21-hydroxylase deficiency represents 90-95% of all CAH cases. Classic 21-hydroxylase deficiency occurs in 1:5,000 to 1:15,000 live births, while non-classic (heterozygous) mutations of the 21-hydroxylase gene occur in approximately 1 in 60 live births.36
a. CAH classic form :
The CAH classic form affects 1:10,000 to 1:20,000 Caucasian neonates and is classified as two types: salt wasting and simple virilizing:
Salt-wasting CAH: is the most severe form. It represents 75% of all cases of classic CAH and is associated with a complete loss of enzymatic activity of the 21-hydroxylase (<2% of enzymatic activity). The loss of enzyme function leads to a deficiency in the synthesis of cortisol and aldosterone, causing a hydroelectrolytic disorder that presents as hypovolemia, hyponatremia, and hyperkalemia. Also, there are high blood levels of renine, weight loss or failure to gain weight, seizures, and eventually death if the condition is not diagnosed and treated in a timely manner. Clinical manifestations may present between the 1st and 4th weeks after birth. Timely diagnosis is fundamental for an early and proper management, to avoid morbidity and mortality. This is especially important in males, as they do not exhibit sexual ambiguity, and genital hyperpigmentation may be the only external symptom, along with penile enlargement.16,33,35,38,39 It is important to stress again that newborns with XX karyotype will have various degrees of virilization, as described later in this paper.36
An excess of adrenal androgens during embryonic development results in virilization of external genitalia in 46,XX newborns. This may present as enlarged clitoris, rugate and partially fused labia majora, common urogenital sinus, and even development of a male urethra. Internal genitalia are usually intact.38
Substitutive therapy with glucocorticoids and mineralocorticoids is currently the mainstay treatment. Hydrocortisone is the glucocorticoid of choice in boys and girls, with the purpose of not inhibiting growth. In adults, long-acting glucocorticoids, such as dexamethasone and prednisone, may be used. Mineralocorticoid substitution is given with fludrocortisone, with the dose adjusted to maintain the plasma renin activity in the normal mid-range.6,36,38 Lack of treatment and excessive androgens may lead to several problems: progressive virilization in girls; premature development of secondary sexual characteristics in both males and females with bone age advancement and central precocious puberty. Consequences may include early epiphyseal cartilage closure, resulting in short stature.38
Simple virilizing CAH: Simple virilizing CAH accounts for 25% of classic CAH cases. The 21-hydroxylase activity is higher in this form than in the salt-wasting form. The accumulation of adrenal steroid precursors results in androgen hyperproduction and the consequent development of ambiguous genitalia and various degrees of virilization in girls. Since there is adequate aldosterone production, salt waste does not occur. Diagnosis may be delayed.35
b. Non-classic CAH:
In non-classic CAH, 21-hydroxylase activity is higher than in the classic form (20-50% of the normal enzymatic function). Cortisol and aldosterone levels are sufficient to maintain vital functions, but they are not enough to inhibit ACTH secretion by negative feedback, leading to adrenal hyperplasia and hyperandrogenism. At birth, girls have normal external genitalia and, usually, normal levels of 17-OH-progesterone.35
Diagnosis is usually belated. In boys and girls, symptoms usually appear after age 5. These include pubarche (a sign of peripheral precocious puberty), acne, growth acceleration, and advanced bone age. According to a multicentric study by Moran et al. in 2000, the most common symptoms in adults are hirsutism (60-78% of cases), menstrual cycle disorders (55%), acne (33%), and reduced fertility (12%). These symptoms are more pronounced in females than in males. For males, in fact, the disease is usually asymptomatic or produces manifestations such as acne or reduced fertility.38
2.1.1.2. 11β-hydroxylase deficiency (CYP11Β1)
The 11β- hydroxylase (11OHD) deficiency represents 5-8% of CAH, and it occurs in 1:200,000 live births. This deficiency is caused by mutations in the gene of the 11β-hydroxylase located in chromosome 8q21. The enzyme catalyzes the conversion of 11-desoxycortisol in cortisol and of 11-desoxicorticosterone in corticosterone.2 The enzyme deficiency also has two possible phenotypes, classic and non-classic, depending on clinical severity and percentage of enzymatic activity loss.37
The 11β- hydroxylase deficiency causes a reduction of cortisol secretion, along with an accumulation of 11-desoxycortisol and 11-deoxycorticosterone. While this combination avoids early salt waste, it later results in hypertension. Increased production of adrenal androgens also causes virilization of external genitalia in newborn females. Also, a non-classic form of 11OHD has been described in female patients born with normal genitalia. These cases develop signs of androgen excess during childhood or adult life.37
Phenotypically, there is virilization of external genitalia in females and peripheral precocious puberty in males. Two-thirds of 11βOHD cases show hypertension at the moment of the diagnosis. The non-classic form of 11βOHD has clinical characteristics similar to the non-classic forms of 21OHD, with no abnormalities at birth. Patients may seek medical attention due to mild virilization, peripheral precocious puberty, hirsutism, or menstrual irregularity. Hypertension is not found in this non-classic form of 11βOHD deficiency.38,40
2.1.1.3. Type 2 deficiency of 3β-hydroxysteroid dehydrogenase (HSD3B2)
Type 2 deficiency of 3β-hydroxysteroid dehydrogenase is one of the least common forms of CAH (less than 2% of cases). The 3βHSD catalyzes the conversion of Δ5 steroids (pregnenolone, 17-hydroxypregnenolone, and DHEA) to Δ4 steroids (progesterone, 17OHP, and androstenedione, respectively). This affects the three biosynthetic paths (mineralocorticoids, glucocorticoids, sex steroids).2,38 It alters steroidogenesis in the adrenal glands and in the gonads, producing salt waste in both sexes, incomplete masculinization of external genitalia in males, and clitoral growth in females. Some females, though, may have normal external genitalia. In females, the non-classic phenotype is characterized by hirsutism and menstrual irregularities.2,37
2.1.1.4. P450 oxidoreductase (POR) deficiency
The POR deficiency is a rare type of CAH. A review of 2017 literature described only 100 cases documented worldwide.41 The POR deficiency is caused by mutations in the POR gene (7q11.2) which encodes an enzyme that transfers electrons from the NADPH to certain enzymes involved in steroidogenesis, including 17-α-hydroxylase (CYP17A1), 21-Hydroxylase (CYP21A2), and placental aromatase (CYP19A1). The mutation results in a partial deficiency of these enzymes. High levels of 17-hydroxyprogesterone are found, though not as high as in the 21-hydroxylase deficiency. A prenatal excess of androgens causes virilization of external genitalia, though postnatal androgen levels may be normal or low. Usually, there is no mineralocorticoid deficiency. The ACTH values may be high.2,37 The POR deficiency may cause ambiguous genitalia in both sexes. In males, this is due to insufficient synthesis of testosterone and dihydrotestosterone and, in females to the interrupted conversion of androgens into estrogens.42
Maternal virilization occurs during pregnancy in 40.8% of cases due to deficiency of placental aromatase (CYP19A1). Virilization in the mother may present as acne, hirsutism, and voice deepening, which revert after pregnancy ends.43
The literature reports that 75% of individuals with POR deficiency present ambiguous genitalia, 82.7% present skeleton abnormalities similar to those in Antley-Bixler syndrome (craniofacial malformations, craneosynostosis, arachnodatilia, clinodactilia, radiohumeral sinostosis, and arched femurs), 74.6% have adrenal insufficiency, and 46.7% of female patients have ovarian cysts.42
2.1.2 Mutations of the glucocorticoid receptor (GR) gene (generalized glucocorticoid resistance syndrome)
This infrequent condition is characterized by increased cortisol secretion without clinical evidence of biological hypercortisolism, defined as high free cortisol in urine and morning cortisol levels > 50 nmol/l after a suppression test. Patients have increased ACTH secretion, which results in elevated androgen and/or mineralocorticoid production.44 Age of symptoms onset varies from birth to adulthood, according to severity of clinical presentation. This presentation is variable and broad, ranging from completely asymptomatic to severe hyperandrogenism, fatigue and/or hypertension, with or without electrolytic abnormalities, and even death. The most severe loss-of-function GR mutations have been seen in children, some with ambiguous genitalia at birth, hypertension with small brain hemorrhagic infarcts, or sepsis.2,44
Generalized glucocorticoid resistance syndrome has been associated with GR gene (NR3C1) loss-of-function mutations, although there are rare cases in which no variants were identified in this gene.45 Also, some polymorphism in NR3C1, as well as in ER22/23EK (rs6189 and rs6190) and GR-9β (rs6198) have been linked to this syndrome . Data, however, seem inconsistent.44
2.2.1 Aromatase deficiency (CYP19Α1)
This rare disorder is caused by mutations in the CYP19 gene (15p21.1) that encodes the aromatase (P450arom) enzyme, which catalyzes the conversion of androgens into estrogens and is mainly found in the ovary, placenta, breast glands, and bone.
Phenotypically, it leads to maternal virilization during pregnancy and ambiguous genitalia in the female fetus. The androgen levels are elevated, and estrogen levels usually are undetectable at birth. In later stages, the deficiency is associated with lack of mammary development, primary amenorrhea, tall stature, and polycystic ovaries. There is some phenotypical variation with unusual presentations, such as genital ambiguity at birth with some degree of mammary development at puberty, or with minimal androgenization at birth but no puberty. Those findings suggest that residual aromatase activity may be enough for mammary and uterine development at puberty.46
2. 3. Maternal
There are maternal causes for external genitalia masculinization in female fetuses including androgen-producing tumors such as the adrenocortical virilizing, ovarian, or Krukenberg tumor, an ovarian metastasis of a primary tumor from abdominal or retroperitoneal organs. Another androgen-producing lesion is the pregnancy luteoma, a benign, hormone-dependent, ovarian tumor that occurs during pregnancy and reverts after delivery. It is usually asymptomatic, and phenotypically it may cause maternal masculinization and, in girls, various degrees of external genitalia virilization.2
3. Other abnormalities
There are other disorders related to 46,XX DSD, such as vaginal atresia, cloacal exstrophy, uterine abnormalities (Müllerian agenesis/hypoplasia), and labial adhesions. All may be isolated or part of a syndrome.1,2
The Mayer-Rokitansky-Küster-Hauser (MRKH) is a representative syndrome, with an incidence of 1:4.500 live female births. It is characterized by aplasia of the uterus and the upper part of the vagina in individuals with normal 46,XX karyotype and normal ovarian function and with normal secondary sexual characteristics at puberty. There are usually no symptoms during childhood, and the first manifestation is primary amenorrhea.47
There are 2 phenotypic types: 1) Rokitanksy sequence or type I MRKH syndrome is an isolated utero-vaginal aplasia with normal Fallopian tubes. 2) The MURCS association or type II MRKH syndrome is an incomplete uterine aplasia with aplasia/hypoplasia of one or both tubes. It may include other malformations, such as renal abnormalities (unilateral agenesia, ectopia, or horseshoe kidney), skeletal, cardiac, and digital abnormalities, and hearing impairment.
The etiology of the MRKH syndrome remains unknown, although several candidate genes have been proposed, among these WT1, PAX2, HOXA7,HOXA13, and PBX1.47
Other disorders have been associated with mutation in specific genes. One is WNT4, where variants in 46,XX females have been associated with a syndrome characterized by an absence of Müllerian derivatives and by hyperandrogenism. The condition may present renal abnormalities.47
Conclusion
The DSD are conditions characterized by abnormality in gonadal, phenotypical, and anatomical sex development. Due to multiple etiologic factors and the great variability of phenotypical presentations, diagnosis is usually difficult and requires multilevel analysis performed by multidisciplinary team (endocrinologists, urologists, geneticists, psychiatrists among others).
Etiological study has identified several genes involved in the embryonic development of gonads. Alterations in those genes may alter embryonic processes of sexual determination and differentiation, giving rise to various DSD. Any genetic or environmental factor that alters the processes of sexual determination or differentiation may result in a DSD, which will have phenotypical manifestations depending on the affected genes or pathways.
In the light of current knowledge, several genes involved in the male gonadal differentiation and determination pathway have been well identified and described, however, there is still a large field to be explored regarding the number of genes involved in the female gonadal differentiation pathway. This limitation in knowledge generates difficulties in the search of the possible molecular causes of these disorders.
Additionally, the great variability of phenotypical presentations in DSD 46, XX, along with the lack of knowledge of the pathophysiology of this diseases, generate confusion when trying to focus the diagnosis. This contributes to the delays and inadequate managements that results in a detriment in the health and quality of life of the patients.
More studies are necessary, specially focused in the identification of genes related to specific disorders of gonadal development, that could help understand in a broader way the physiopathology of these conditions.

