











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Biomass and biofuels in particular are nowadays considered as promising and sustainable alternative fuels due to their renewability, reduced toxic-gas emissions, and biodegradability. Direct use of vegetable oils in diesel engines has not been entirely successful due to poor properties such as low volatility, reactivity of the unsaturated molecules, and high viscosity, which can lead to coking on engine injectors, carbon deposits, and plugged orifices during prolonged engine performance. Transesterification reaction is the most often used method to improve these properties.
Any triglyceride feedstock present in the vegetable oils and animal fats (including algal lipids) can be transesterified to produce the commonly named biodiesel, which is defined as an oxygenated fuel comprised of monoalkyl esters of long chain fatty acids and is designated by the American Society of Testing Materials (ASTM, 2005) as B100.
The most common alcohol used in biodiesel production is methanol, and it is therefore said that biodiesel is comprised of a mixture of fatty acid methyl esters (FAMEs). In addition, FAME distribution in biodiesel depends directly on fatty acid profiles that were present in the source material employed in their synthesis. The most common fatty esters contained in biodiesel (Figure 1) are those obtained from palmitic, stearic, oleic, linoleic and linolenic acid. This holds for most biodiesel feedstocks, such as soybean, sunflower, canola, palm and peanut oils (Knothe, 2010).
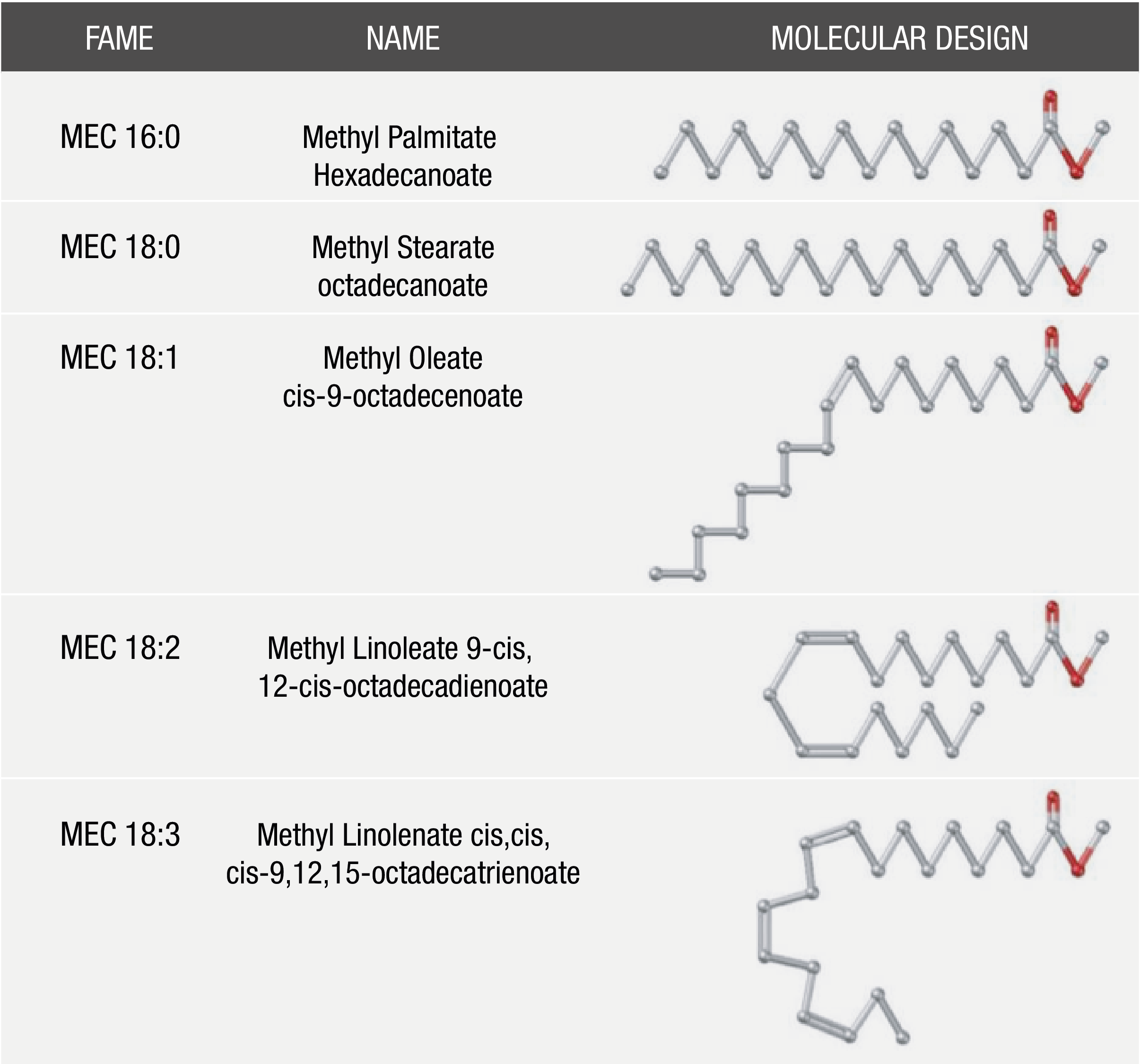
Figure 1 Predominant fatty acid methyl ester (FAME) compounds in the most common biodiesel feedstocks.
Several studies have been aimed at establishing a heuristic relation between fuel properties and fatty acid molecular structure and composition (Schönborn, Landommatos, Williams, Allan, & Rogerson, 2009). Accordingly, the variability of the biodiesel feedstock (presence or absence of any FAME as well as the amount) will generate differences in biodiesel molecular structure. These differences lead to distinctive patterns in fuel energy release rates and pollutant formation. The experimental measure of such properties for all types and possibilities of biodiesel fuel blends is both impractical and expensive. The experimental data needed to develop thermodynamic models are, however, very scarce for complex and large molecules such as biodiesel ones, and experimental measures are extremely costly and time consuming. Thermodynamic models afford a simple and immediate way to predict the physical properties of biodiesel fuels (Csernica & Hsu, 2011; Huber, Lemmon, Kazakov, Ott, & Bruno, 2009; Olivera, Ribeiro, Queimada, & Couthinho, 2011; Olivera, Teles, Queimada, & Coutinho, 2009). However, we consider that a molecular-based cubic equation of state (EOS) is necessary for a better approach to thermodynamical properties.
The Statistical Associating Fluid Theory (SAFT) approach (Chapman, Gubbins, Jackson, & Radosz, 1990; Perdomo, Perdomo, Millán, & Aragón, 2014) provides an EOS that takes into account the anisotropies of the fluid, both due to molecular shape and directional forces. The SAFT-VR approach (Gil-Villegas, Galindo, Whitehead, Jackson, & Burgess, 1997), wherewith fluids are modeled as chains of tangentially bonded hard spheres interacting with potentials of variable range, has also been used to model the vapor-liquid equilibria of three FAME biodiesel compounds (Perdomo & Gil-Villegas, 2010), modelling each FAME as a chain of spherical segments that can associate due to the presence of short-ranged attractive sites. These sites, as well as the intermolecular interaction among three monomer segments, were modeled by square-well potentials of variable range.
The same approach was implemented to model biodiesel blends using a corresponding mixing rule in the SAFT-VR approach and a cross-binary parameter among species for the unlike energy parameter, making it possible to determine isochoric heat capacities, speed of sound as well as the phase diagram of binary and ternary FAME mixtures (Perdomo & Gil-Villegas, 2011). In spite of the aforementioned rigorous approaches, classical EOS have been successfully applied in the chemical industry to describe the thermodynamic properties of a wide range of pure fluids and mixtures. Although it is known that classical EOS fail in the description of second-derivative thermodyanamic properties and are thus usually applied only for phase equilibrium calculations, their simplicity and wide applicability have justified continued efforts in their improvement.
In this study we propose a model based on the classical Peng-Robinson equation of state (PR-EOS) (Peng & Robinson, 1976) in order to evaluate the departure thermodynamic expressions (first- and second-derivative properties) in biodiesel fuels. To achieve this goal, the herein developed approach uses two concepts that have been commonly applied to the PR-EOS: the temperature dependent function α(T) and the translated volume. In the first case an exponential α(T) function, proposed by Gasem, Gao, Pan, and Robinson (2001), was implemented in order to improve vapor pressure prediction in pure FAMEs. It is important to highlight that the selection of the temperature function was the result of a systematic search among a wide range of correlations reported in the literature. The translated volume concept was implemented due to the fact that second-order thermodynamic properties need accurate densities and derivatives with respect to the total volume, and the original α(T) function has no significant influence on the description of volumetric properties.
Materials and methods
Model development
The methodology used for modeling biodiesel systems is based on the classical volume-translated PR-EOS (VTPR-EOS) (Equation 1) (Ahlers & Gmejling, 2001; González-Pérez, Coquelet, Paricard, & Chapoy, 2017; Peng & Robinson, 1976).
Where
and
T c , P c , R, and v are the critical temperature, the critical pressure, the universal gas constant, and the molar volume respectively. The translation parameter c is defined as the difference between calculated and experimental volume at the considered reduced temperature (T r ) and is obtained via the Rackett equation (Rackett, 1970):
where Z c = P c / Rρ c T c is a critical compressibility factor. Constants c 1 , c 2 , and c 3 have to be optimized for a particular biofuel mixture. The α-function is a temperature-dependent model function that has been the subject of several studies in order to improve the precision of EOS. Many proposed α-functions use either a high-order polynomial in the acentric factor or temperature to correlate vapor pressures more precisely. For this study, the α-function proposed by Gasem et al. (2001) was selected:
where ω is the acentric factor that characterizes the non-sphericity of molecular interactions. Constants A, B, C, D, and E also have to be optimized for the considered biodiesel compounds.
In order to estimate ω, the group contribution method proposed by Constantinou, Gani, and O’Connell (1995), which does not need other primary properties for its determination, was implemented. Table 1 shows the primary and secondary groups needed to represent FAME biodiesel constituents. Once the framework is settled (groups and occurrences), the ω is calculated as follows (Constantinou et al., 1995):
Table 1 Group contribution information for estimating the acentric factor of the five biodiesel compounds considered in this work.
| Group | ω 1i,2j | MEC 16:0 | MEC 18:0 | MEC 18:1 | MEC 18:2 | MEC 18:3 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ni | Mj | Ni | Mj | Ni | Mj | Ni | Mj | Ni | Mj | ||
| First order | |||||||||||
| CH3 | 0.29602 | 2 | 2 | 2 | 2 | 2 | |||||
| CH2 | 0.14691 | 13 | 15 | 13 | 11 | 9 | |||||
| CH=CH | 0.25224 | 0 | 0 | 2 | 4 | 6 | |||||
| CH2COO | 0.75574 | 1 | 1 | 1 | 1 | 1 | |||||
| Second order | |||||||||||
| CH2-CHm=CHn | -0.0115 | 0 | 0 | 1 | 2 | 0 | |||||
Source: Constantinou et al. (1995).
where a ω = 0.4085, b ω = 0.5050, and c ω = 1.1507 are universal constants, ω 1i is the contribution of the type i first-order group which occurs N i times and ω 2j is the contribution of type j second-order group, which occurs M j times. Both contributions are involved, A ω = 1 (Constantinou et al., 1995).
Fitting data
The complete parameter set θ ≡ (A, B, C, D, E, c 1 , c 2 , c 3 ) was determined for each of the most commercially-used FAMEs (Table 2) (Hoekman, Broch, Robbins, Ceniceros, & Natarajan, 2012), of which methyl palmitate (MEC 16:0) and methyl stearate (MEC 18:0) are saturated, and methyl oleate (MEC 18:1), methyl linoleate (MEC 18:2) and methyl linolenate (MEC 18:3) are unsaturated. For this, the proposed α(T) function was fitted to the theoretical vapor pressure values of the experimental liquid-vapor equilibrium data corresponding to the FAMEs studied by Rose and Supina (1961). The critical region was excluded from the parameter regression.
Table 2 Parameter set θ for the five fatty acid methyl esters considered.
| MEC 16:0 | MEC 18:0 | MEC 18:1 | MEC 18:2 | MEC 18:3 | |
|---|---|---|---|---|---|
| A | 1.9904 | 2.6131 | 2.3646 | 4.4464 | 1.9303 |
| B | 0.9278 | 0.9356 | 0.0043 | 0.5957 | 0.0014 |
| C | 0.0906 | 0.6382 | 0.2285 | 0.6195 | 0.0017 |
| E | 0.4376 | 0.2930 | 0.3522 | 0.9063 | 0.7524 |
| D | 0.0778 | -0.5060 | -0.0027 | -1.1680 | -0.0383 |
| c1 | -0.3921 | -0.2942 | -0.4221 | -0.2302 | -0.2881 |
| c2 | 1.6488 | 1.7821 | 1.3134 | 1.9822 | 1.7439 |
| c3 | 0.0981 | 0.0146 | 0.0154 | 0.0321 | 0.0048 |
In classical optimization procedures, the relevant parameters are optimized either for liquid-vapor equilibrium or second-derivative properties, but not both simultaneously. Due to the known difficulty to calculate second-derivative properties by classical EOS, because of their direct dependence on the molecular structure of the fluid, the following relative least square objective function was used (Liang, Maribo-Mogensen, Thmosen, Yan, & Kontogeorgis, 2012):
where N
e
are the number of experimental data points for vapor pressures
To determine an optimal multiparameter set, a hybrid minimization method was implemented, combining global-search and local-search algorithms. Additionally, an iterative procedure was used to obtain the most adequate values: 1) the values of the pure component parameters were estimated with the original values (c 1 , c 2 , c 3) proposed by Dietrichs, Rarey, and Mehling (2006), 2) a regression was performed on the coefficients A to E in the modified α(T) function, 3) the pure component parameters were estimated with new values (c 1 , c 2, c3), and 4) steps 1 to 3 were repeated until the desired convergence was obtained.
The simplex method was used to reduce the convergence domain, and then the Levenberg-Marquardt pseudo-second order method (Nocedal & Wright, 2000) was applied until the function gradient attained its minimum value, that is:
In order to verify the optimal parameter fitting, the saturation vapor pressure curves, representing P vs T and the Clausius-Clapeyron model, were constructed for each FAME.
Modeling thermophysical properties in biodiesel systems
This section presents the expressions and numerical procedures for calculating the key properties required in the study of fuel quality, combustion, and injection system design in engines.
Boiling point: at atmospheric pressure and called normal boiling point, it is the basis for predicting the critical properties and those dependent on temperature, such as vapor pressure, density, latent heat of vaporization, viscosity, and surface tension, which are required for biodiesel combustion modeling (Yuan, Hansen, & Zhang, 2003), but the wide temperature ranges required for these properties make it difficult to experimentally measure them. Therefore, the accurate boiling points of the pure fatty acid esters and their biodiesel mixtures are useful for predicting fuel properties and are consequently important for combustion modeling.
Vapor pressure in pure biodiesel compounds: a simple procedure can be implemented in order to satisfy the liquid-vapor equilibrium criteria and estimate the vapor pressure at some constant temperature. First, it is necessary to estimate an initial approximation of the pressure value P k with a lower value than the critical pressure, and then recalculate it using the recurrence relation:
where ϕ is the fugacity coefficient. Further details are given in Appendix 1.
The vapor pressure in the fuel mixtures is also useful to assess the risk level in the different biodiesel production stages, as well as in its storage and transport.
Liquid heat capacities: it is a measure of the amount of energy required by a mol (or mass unit) of a substance to raise its temperature by a unit degree. It is of great importance in the design of industrial processes and engineering calculations (i.e., distillation, heat exchanger designs, estimation of reaction enthalpies as a function of temperature, thermodynamics of combustion analysis, etc.). Ignition and combustion in diesel engines are strongly influenced by the fuel spray, and this in turn is affected by heat capacities and heat of vaporization.
The heat capacities (Equation 10) of biodiesel systems can be calculated by two methodologies (Diedrichs, Rarey, & Mehling, 2006). The first one proceeds from the thermodynamic expressions that give a relation between measurable quantities and the entropy after applying the appropriate relations:
where
Speed of sound and isentropic bulk modulus. Differences in physical properties between biodiesel and petroleum-based diesel fuel may change the engine's fuel injection timing and combustion characteristics (Schönborn et al., 2009; Tat & Van Gerpen, 2003) by altering both the combustion timing and rate. The properties that have the greatest effect on fuel injection timing are the speed of sound (v s ; Liang et al., 2012) and the isentropic bulk modulus. In this work the values of the aforementioned key properties of biodiesel components and blends at temperature and pressure ranges most commonly encountered in injection system conditions are predicted.
Once v s is obtained, the isentropic bulk modulus (β) is readily given by the following expression (Tat & Van Gerpen, 2003):
Multicomponent biodiesel system
In order to predict key physico-chemical properties in FAME mixtures, the classical one-fluid theory mixing rules defined by the following set of equations was implemented (Diedrichs et al., 2006; Peng & Robinson, 1976):
where
The coefficients a i , b i , and c i are calculated from Equations 2, 3 and 4, and x i is the molar fraction in the liquid phase of component i in the considered mixture. To calculate vapor-liquid equilibria, specifically the bubble (or dew) point pressure (P*), the fugacity coefficient for component i in a FAMEs mixture is written as (Peng & Robinson, 1976):
The molar fraction in the vapor phase of component i is expressed in terms of T, P*, and x i as:
where к i is the distribution coefficient denoted by:
The P* was obtained through a conventional Rachford-Rice procedure, without any assumption about the ideality in the FAME mixtures, which has been indeed made in several investigations (Yuan, Hansen, & Zhang, 2005); for this, the following equation was solved:
where ψ represents the vaporized fraction in the system, which can take any value between 0 to 1. The iteration procedure was started by estimating the bubble point as:
where P i sat is calculated with the procedure explained in Appendix 1.
All calculations were done in MatLab language Ver. 8.5 (Math Works Inc. Natick, Massachusetts, USA).
Results and discussion
Table 3 shows the parameters of the
Clausius-Clapeyron relation (
Table 3 Clausius-Clapeyron relation parameters of fatty acid methyl esters (FAMEs) for the biodiesel compounds studied in this work, compared with the experimental data of Scott et al. (1952) with the corresponding deviation percent.
| FAME | α1 | γ | r2 | Experimental data | Calculated data | Deviation percent |
|---|---|---|---|---|---|---|
| MEC 16:0 | 9.3431 | -4,199.03 | 0.999 | 80,219.1 | 73,562 | 1.1 |
| MEC 18:0 | 9.5402 | -4,496.95 | 0.999 | 85,703.8 | 74,673 | 5.7 |
| MEC 18:1 | 9.3945 | -4,395.05 | 0.997 | 84,446.8 | 76,077 | 7.4 |
| MEC 18:2 | 9.4120 | -4,397.86 | 0.999 | 83,610.4 | 77,697 | 7.7 |
| MEC 18:3 | 9.5566 | -4,472.14 | 0.999 | 84,573.4 | 73,800 | 7.2 |
1α and γ: Clausius-Clapeyron constants.
Table 4 Values of the constants cp0 to cp7 for the fatty acid methyl esters.
| MEC 16:0 | MEC 18:0 | MEC 18:1 | MEC 18:2 | MEC 18:3 | |
|---|---|---|---|---|---|
| cp0 | 120.529 | 247.115 | 90.2385 | 190.986 | 79.5913 |
| cp1 | 0.0801627 | -0.0916606 | 0.14118 | 0.020213 | 0.214648 |
| cp2 | 345.62 | 276.94 | 234.797 | 437.371 | 290.379 |
| cp3 | 2,952.37 | 556.17 | 613.529 | 3,052.11 | 1,213.24 |
| cp4 | 289.038 | 408.997 | 335.768 | 287.222 | 81.4323 |
| cp5 | 734.653 | 1,311.85 | 1,405.31 | 746.631 | 578.752 |
| cp6 | 301.639 | 472.702 | 431.66 | 321.956 | 474.881 |
| cp7 | 1,593.55 | 2,825.71 | 2,867.76 | 1,624.33 | 2,799.79 |
Source: Huber et al. (2009).
Table 5 Gas-phase standard enthalpies at 298.15 K for the biodiesel compounds studied in this work.
| FAME | (hv (Kcal·mol-1) |
|---|---|
| MEC 16:0 | -167.8 |
| MEC 18:0 | -177.3 |
| MEC 18:1 | -147.2 |
| MEC 18:2 | -116.1 |
| MEC 18:3 | - 85.6 |
FAME: fatty acid methyl esters. Source: Osmond et al. (2007).
A good prediction in the vapor pressure and, consequently, in the heat of vaporization, is also necessary in order to have reasonable accuracy in second-derivative properties because it is directly related to the change in heat capacity in the vaporization region (Equation 20).
It is important to mention that the herein proposed model is the result of a systematic testing of various α(T) functions previously reported in the literature and reviewed in Gasem et al. (2001). They found good approximations for the vapor pressure data obtained. However, they did not obtain a similar agreement for second-derivative properties, where high deviations in the speed of sound were remarkable.
Figure 2 presents the speed of sound prediction for the five considered FAMEs when only the α(T) proposed by Soave is implemented without any volume correction. As can be readily seen, the predicted values are in strong disagreement with the experimental ones, in spite of good prediction in the vapor pressure. Additionally, for saturated FAMEs, the presence of double bonds in the molecular structure renders the prediction very difficult, especially at low temperatures.
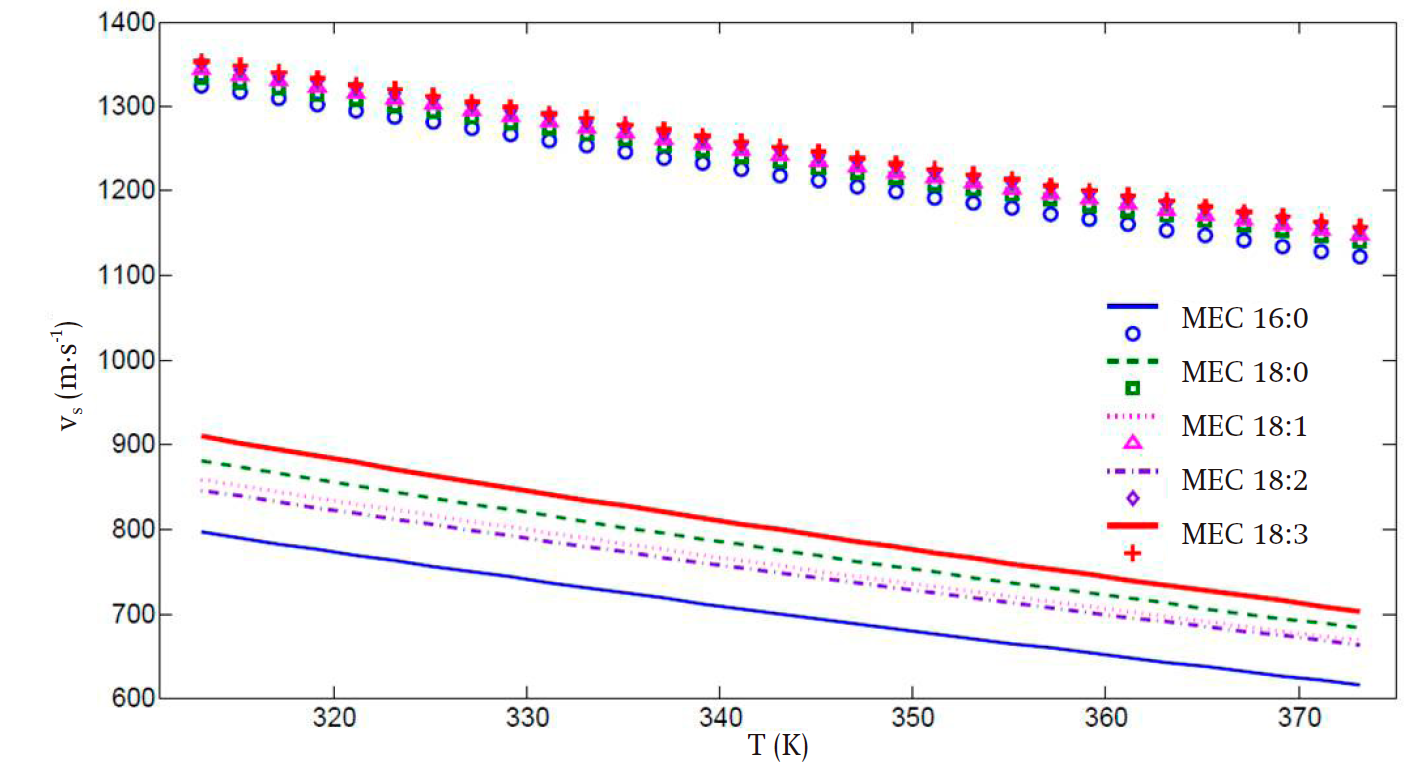
Figure 2 Predicted speed of sound (vs) (lines) for all fatty acid methyl esters employing the Soave α(T) function.
In order to address the aforementioned issues, the volume-translated modification was introduced to generate dependence in the vapor pressure curve with respect to the volume ((P/(ρ), thus obtaining an agreement in both sets of variables (single and double derivatives) without losing predictive capability in the prediction of vapor pressure values.
Figures 3 and 4 present the predicted speed of sound and bulk modulus values, respectively. In spite of the deviations, especially in the high temperature regime, the improvement in the calculation of these properties is remarkable when the translated volume modification is incorporated, in comparison with those presented in Figure 2, with the advantage that the previously obtained parameter set compensates the predictions of the liquid vapor equilibrium and second-derivative properties. Thus, the key properties of the biodiesel fuel components can be predicted with the same set of values for all temperature and pressure regimes considered. This is a practical advantage of our proposal worth stressing.
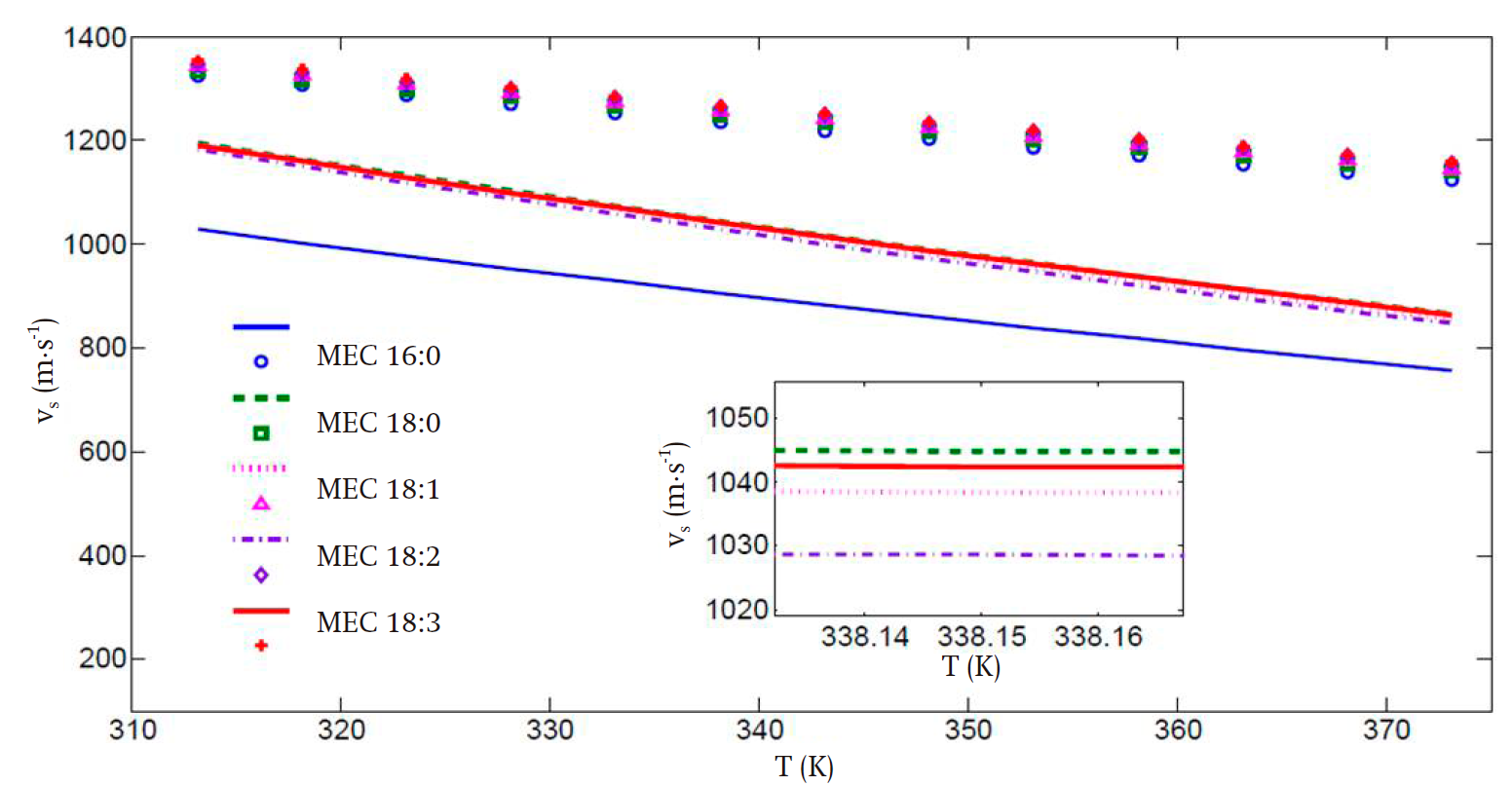
Figure 3 Speed of sound (v) vs temperature (T) calculated for all considered fatty acid methyl esters, using the volume translated parameters. The inset presents the same data, but on a finer scale, in the high temperature region.
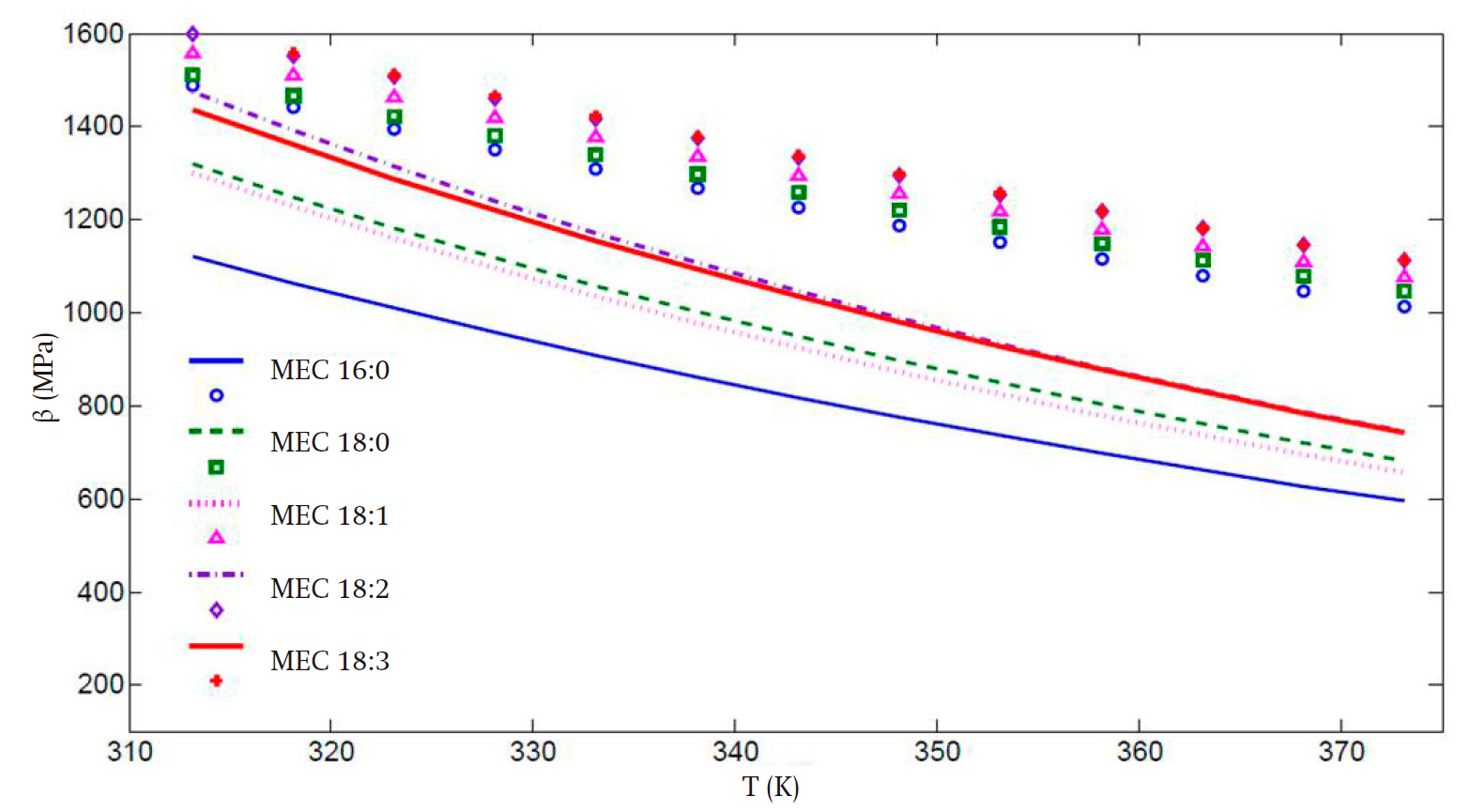
Figure 4 Isentropic bulk modulus (β) vs temperature (T) calculated for all fatty acid methyl esters, using the volume translated parameter.
It can be seen that, for the speed of sound, the average deviations of the theoretical values from the experimental ones for the FAMEs with high carbon number are of 14 and 21 % in the low and high temperature regimes, respectively. These results are clearly superior compared to those in Figure 2, wherein the corresponding deviations in the low and high temperature regimes are of 35 and 38 %. Also, in Figure 3, a more uniform behavior is obtained for all FAMEs with high carbon numbers.
For the bulk modulus the aforementioned behavior is not observed, and there is an increasing deviation, from 17 to 29 %, of the theoretical values for all FAMEs as temperature increases.
Once the optimum parameter set values were determined, the properties of real commercial biodiesels were calculated. For this, a single Van der Waals one-fluid theory mixing rule (Equations 13 and 14) was implemented to take into account the mixing effects when dealing with a multicomponent FAMEs system. Biodiesel from palm, soy and lard was analyzed; its composition was taken from Goodrum (2002), and the P* results are reported in Figure 5, where the good agreement between our predictions (once the Rachord-Rice criteria are incorporated, Equation 18) and the experimental data taken from Goodrum (2002) can be readily seen. According to these results, it can be inferred that a single simple mixing rule is sufficient to consider the mixing effects in long-chained methyl esters.
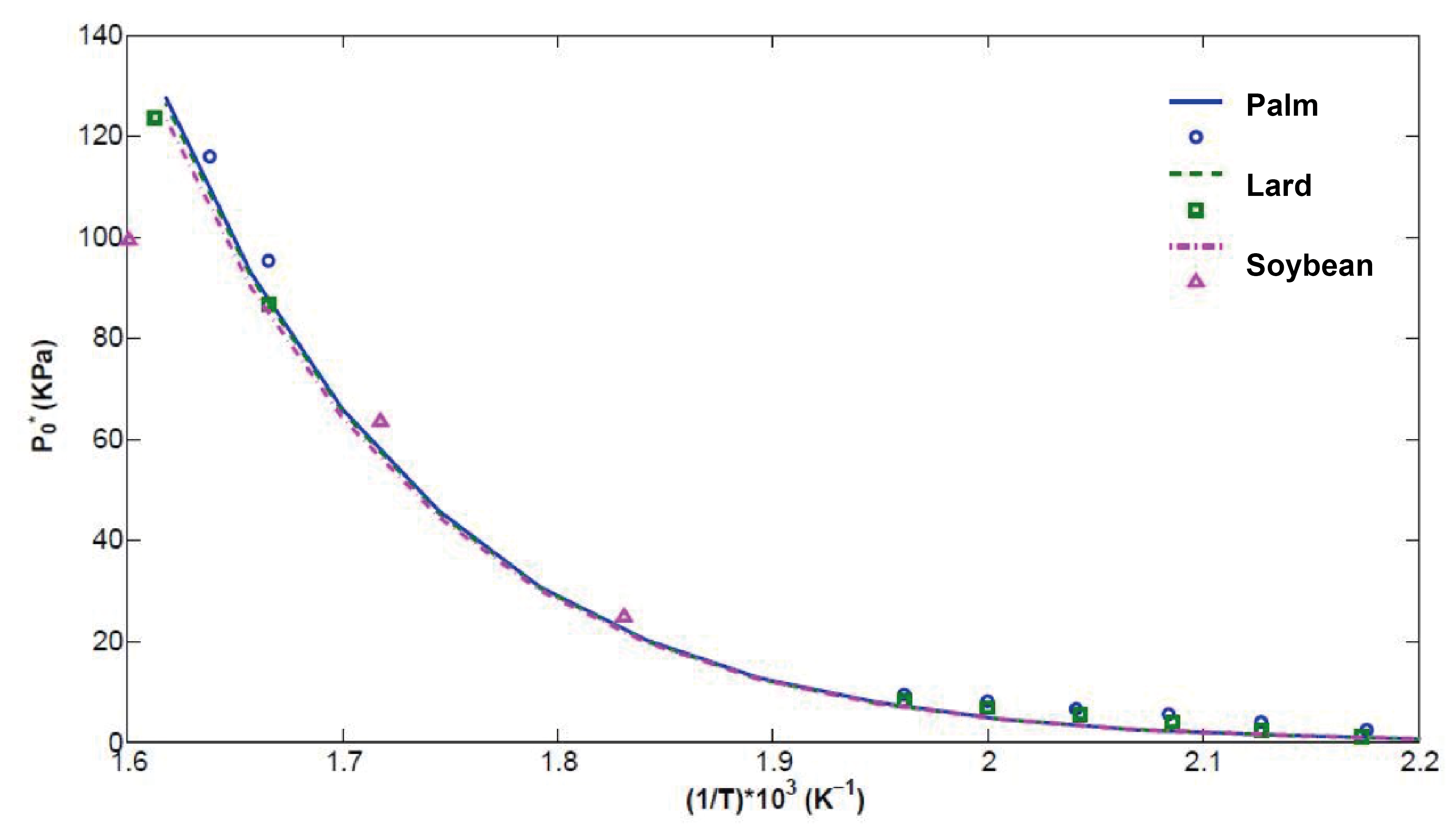
Figure 5 Predicted (lines) bubble point values vs inverse temperature (1/T) for the commercial biodiesels obtained from palm oil, lard and soybean oil. Circles and squares correspond to predicted values from the Antoine equation reported in Yuan et al. (2005), and triangles to experimental values reported by Goodrum (2002).
The proposed model attains an appreciable improvement in the prediction of second-derivative properties like heat capacity (isochoric and isobaric), bulk modulus and speed of sound. It is important to remark that the accuracy of the prediction of liquid-vapor properties is kept with the same set of fitted parameters. In addition, the model depends on very limited experimental information (only pure FAMEs critical properties) and does not need other primary properties for its determination. Therefore, it can become a valuable tool for both the design of biodiesel injection systems and the interpretation of the combustion process.
Conclusions
The obtained results reveal that classical EOS with only a single α(T) modification cannot describe with acceptable accuracy both kinds of physico-chemical properties. However, fitting pure component parameters can significantly improve the speed of sound and bulk modulus predictions if experimental data are taken into account in the objective function to be optimized.
In the case of FAME mixtures (real biodiesel systems) a single mixing rule was sufficient to obtain very good agreement with experimental results already reported in the literature. Thus, in this work we have presented a model of predictive nature, based on physical principles, that allows the evaluation, with the same parameter set, of all physico-chemical properties necessary to assess the performance and quality of biofuels in processes such as atomization and combustion in diesel engines. In general, the proposed method turns out to be good for static properties but it is not so easy to apply for dynamic properties. However, we consider it a good approach since it affords a way to obtain a qualitative agreement of the speed of sound with available experimental data.

