











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
As the cost of genotyping continues to decrease, it has been met with an increase in demand amongst farmers. Producers recognize that animal genetic resources must be preserved because of their contribution to human livelihood, now and in the future1. The level of inbreeding within one’s herd has become of particular concern due to selection efforts and has thus been a large motive behind genotyping. An increase in inbreeding leads to a variety of negative effects, such as reduction in phenotypic values for some traits, of genetic variance, and higher frequency of homozygous genotypes2.
Therefore, the desire to manage this coefficient has increased, especially amid small, local herds or dairy stock. Pedigrees may not always be available or sufficient to calculate inbreeding (F PED) within one’s herd, but the coefficient of inbreeding could effectively be determined from genotypes as a means of heterozygosity within the population (F IS) or as a measure of whole genome inbreeding from runs of homozygosity (ROH) (F ROH). In fact, several studies in cattle have effectively shown that ROH are suitable to estimate genomic inbreeding coefficients in the absence of a pedigree3-5. Knowledge of runs of homozygosity alone provides not only new possibilities to manage inbreeding in livestock species, but could be used for optimal allocation of resources and maintenance of genetic variation in intensely selected bovine breeds6. Nevertheless, hesitation still exists among farmers to invest in high-density single nucleotide polymorphism (SNP) chips, so preference has lately been given to the low-density chips, as they are more cost-effective when wanting to genotype a large number of animals. The design of these chips has greatly improved and has led to greater gains in reliability and improved readability among SNP genotypes7.
Another large reason for genotyping among farmers is to obtain the genomic estimated breeding values. In fact, many breeders already embrace this value when purchasing semen7. Therefore, farmers could already achieve higher annual rates of genetic gain through using genomically tested bulls, but it is becoming increasingly popular to capture extra value from genotyping females8. This extra value obtained from genotyping one’s herd could be employed as a managerial tool, as genotypes provide considerably more information that just the captured variants at the disease loci featured on the SNP chip. The benefits of this information range from improving the reliability of genomic selection (of both bulls and heifers) to identifying elite females and the best heifers to become herd replacements. As such producers can obtain a better indication of the value of an animal’s respect to the solely expected genomic breeding value and use it to avoid or manage inbreeding, and of course prevent genetic defects by avoiding mating that would cause deleterious alleles to surface8.
The objective of this study was to show how SNP chips could be used as an effective management tool in respect of herd genetic variability. We particularly emphasize that the effective level of genomic inbreeding determined from genotypes is comparable to that obtained from an informative pedigree, and that SNP genotyping allows to disclose genomic regions under selection pressure in the herd as identified by genes within runs of homozygosity.
Material and methods
Genotyping
Genotypes were provided by the National Friesian Italian Breeds Association (ANAFI) for a total of 44 Italian Holstein-Friesian cattle aged 12 to 15 mo all coming from a unique herd, the University experimental station. All individuals had a marker call rate greater than 98 %. Animals used in this study were part of an experimental population raised at University of Milan’s experimental farm, Azienda Agraria Didattico Sperimentale Angelo Menozzi. Animals were genotyped for 30,125 SNP using the GeneSeek GGP Bovine LD v4 array. After excluding SNPs that were not assigned to a bovine chromosome (BTA) or that were assigned to BTA X or mitochondrial DNA, 27,365 SNP remained.
Principal component analysis (PCA)
Additional information available for each individual included its pedigree, Genomic Productivity, Functionality, Type (GPFT) genomic selection index, and batch of genotyping. The PCA was conducted within SNP with the Variation Suite Golden Helix v8.4 software (SVS) (Golden Helix Inc., Bozeman, MT). The procedure used in SVS considered 20 principal components calculating for each of them a relative eigenvalue; the first principal component (eigenvalue=1.188) was plotted against the second (eigenvalue=0.983).
Inbreeding coefficients
The inbreeding estimate of F IS (or ƒ) was calculated using SVS, defined as 1-(HETOBS/HETEXP). This coefficient is equivalent to Wright’s within-subpopulation fixation index with values in the range of -1 to +1. The whole genome inbreeding coefficients (F ROH) were calculated according to9:
Where: L ROH: is the total length of all ROH in the genome of an individual while.
LAUTO: refers to the specified length of the autosomal genome covered by SNP (2,505,649,802 bp in the current study).
Pedigree-based coefficients of inbreeding (F PED) were derived from Pedigree Viewer10. The pedigree included records for 2,760 individuals up to 19 generations as calculated by Pedigree Viewer that orders genealogical information in discrete generations. F PED and F ROH were compared using linear regression and Pearson’s correlation coefficient to test their similarity and to highlight differences in the two methods of estimation in a group of animals of the same herd.
Runs of homozygosity
Runs of homozygosity were estimated for each individual using SVS v8.4. This program does not rely on sliding windows to identify ROH, but instead the algorithm works continuously across an entire chromosome by examining every possible run for a match with the specified criteria11. The following criteria to define ROH were used: 1) Two missing SNP were allowed; 2) One heterozygous genotype was allowed; 3) The minimum number of SNP that constituted the ROH was set to 30; 4) A minimum length of 1 Mb; 5) A maximum gap between consecutive SNP of 1 Mb. Runs of homozygosity were classified into five classes (<2, 2 to 4, 4 to 8, 8 to 16, and >16 Mb) using the same nomenclature as reported by other authors4,5. The incidence of common runs per SNP was plotted using the Genome Browse tool of Golden Helix, and subsequently aligned to the BTAU 5.0.1 bovine assembly to identify genes consistent with SNP in the top 1% and 5% of runs. The online STRING database classified the network amongst these genes12, while gene ontology (GO) was performed through the Protein Analysis Through Evolutionary Relationships (PANTHER) classification system (Release 13.1), (http://www.pantherdb.org/pathway/).
Results
Principal component analysis (PCA)
Individuals were grouped based on their batch of genotyping (n= 6), sire (n= 18), sire country of origin (n= 6), and class of GPFT (n= 5). The different batches of genotyping did not cluster whatsoever, indicating that the time of genotyping did not affect individual genotypes (data not shown). On the contrary, there was a strong clustering when individuals are identified by their sire and their sire’s country of origin (Figure 1). Sires 1 and 5 are especially distanced from the others, while those originating from Canada, Italy, and the United States tend to group as well, albeit separately by origin. Individuals were subsequently classified by their GPFT value in increments of 500, and no clustering was observed based upon those assigned categories.

Inbreeding coefficients
Wright’s inbreeding coefficient estimate of F IS was calculated for each individual. The majority of animals (n= 31) displayed negative F IS, while a smaller proportion (n= 13) possessed positive coefficients (Figure 2). The highest and lowest F IS values were 0.057 and -0.077, respectively. The average F IS existed at -0.018 shoving an increase in heterozygosity due to an outbreeding mating scheme used by the farmer. The majority of individuals possessed F PED coefficients between 0.050 and 0.070 (n= 31); values ranged from 0.044 to 0.116. The whole genome inbreeding coefficient estimate of F ROH was higher overall than the inbreeding coefficients calculated from pedigree. The average F PED was 0.064, while the mean F ROH >1 Mb equaled 0.083.

Figure 2 Regression of individual inbreeding coefficient. A) Regression of inbreeding calculated by pedigree (F PED) on inbreeding calculated on run of homozigosity (F ROH). R2 = 0.205, red line indicates regression line (F ROH = 0.042 + 0.621 * F PED ). B) Regression of F PED on F IS. R2 = 0.214, red line indicates regression line (F is= -0.077 + 0.913*F PED )
Runs of homozygosity
With the ROH criteria set at 1 Mb and minimum of 30 SNP, the SVS software identified a total of 1,950 runs within the population, ranging from 1 to 22 Mb in length (Figure 3). All 44 animals displayed ROH, with the average number of ROH per animal being 44. The largest number of ROH for an individual was 64, while the least number of ROH for an individual existed at 25. The average ROH length was 4.66 Mb, and the greatest amount of ROH existed in the 4-8 Mb range (Table 1). The number of ROH per chromosome was greatest for BTA 10 (122 runs), while lowest for BTA 27 (24 runs). The longest ROH also existed on BTA 10 at over 22 Mb in length, contrary to other results identifying the longest ROH on BTA 813-15. The average number of SNPs falling into a ROH was consistent among ROH length category, ranging from 42 (<2 Mb) to 153 (>16 Mb) SNP. ROH length appeared to be relatively proportional to chromosome size, with longer runs appearing on longer chromosomes.

Table 1 Numbers of ROH per chromosome according to ROH class of length
| BTA | <2 Mb | 2-4 Mb | 4-8 Mb | 8-16 Mb | >16 Mb | Total |
|---|---|---|---|---|---|---|
| 1 | 1 | 20 | 71 | 4 | 0 | 96 |
| 2 | 1 | 31 | 75 | 10 | 0 | 117 |
| 3 | 5 | 22 | 27 | 6 | 1 | 61 |
| 4 | 1 | 37 | 47 | 7 | 0 | 92 |
| 5 | 7 | 36 | 40 | 15 | 0 | 98 |
| 6 | 11 | 48 | 45 | 2 | 0 | 106 |
| 7 | 3 | 45 | 51 | 7 | 0 | 106 |
| 8 | 5 | 32 | 56 | 10 | 0 | 103 |
| 9 | 10 | 23 | 29 | 0 | 0 | 62 |
| 10 | 2 | 43 | 64 | 12 | 1 | 122 |
| 11 | 2 | 36 | 41 | 6 | 0 | 85 |
| 12 | 5 | 14 | 16 | 4 | 0 | 39 |
| 13 | 8 | 32 | 39 | 6 | 0 | 85 |
| 14 | 8 | 50 | 21 | 0 | 0 | 79 |
| 15 | 3 | 20 | 22 | 4 | 0 | 49 |
| 16 | 12 | 25 | 34 | 7 | 0 | 78 |
| 17 | 2 | 17 | 21 | 3 | 0 | 43 |
| 18 | 5 | 21 | 14 | 2 | 0 | 42 |
| 19 | 7 | 30 | 16 | 1 | 0 | 54 |
| 20 | 2 | 24 | 43 | 7 | 0 | 76 |
| 21 | 1 | 24 | 26 | 6 | 0 | 57 |
| 22 | 2 | 20 | 20 | 5 | 0 | 47 |
| 23 | 4 | 16 | 12 | 2 | 0 | 34 |
| 24 | 4 | 16 | 21 | 3 | 0 | 44 |
| 25 | 1 | 22 | 9 | 0 | 0 | 32 |
| 26 | 1 | 21 | 11 | 2 | 0 | 35 |
| 27 | 3 | 12 | 7 | 2 | 0 | 24 |
| 28 | 1 | 13 | 18 | 4 | 0 | 36 |
| 29 | 7 | 18 | 20 | 3 | 0 | 48 |
| Total | 124 | 768 | 916 | 140 | 2 | 1950 |
The genomic regions most commonly associated with ROH were identified by selecting the top 1% and 5% of SNPs most frequently observed (Figure 4, Table 2). The incidence of common runs per SNP indicated that the genomic distribution of ROH was non-uniform across chromosomes. Quantitative trait loci (QTL) corresponding with regions of homozygosity housing the top 5% of SNPs were identified using the Animal Quantitative Trait Loci (QTL) Database (AnimalQTLdb) (Table 2)16.

Figure 4: Incidence of SNPs in ROH identified by SVS. The red and blue lines indicate the adopted threshold for the top 1% and 5% of observations, respectively
Table 2 Genomic regions of extended homozygosity corresponding with the top 1 and 5% of SNP
| BTA | Number of SNP | Start (bp) | End (bp) | Genes, | QTL (https://www.animalgenome.org/cgi-bin/QTLdb/BT/index) |
|---|---|---|---|---|---|
| 1 | 11 | 49891423 | 51147114 | HSF2BP, ALCAM, CBLB | Milk beta-lactoglobulin percentage QTL (108870); Lean meat yield QTL (37225) |
| 1 | 18 | 145059132 | 146790949 | PDE9A, PKNOX1, ITGB2, ADARB1, POFUT2, ICOSLG, TRPM2, SIK1 | Milk protein percentage QTL (105990); Milk C18 index QTL (32646) |
| 2 | 11 | 15361703 | 17142680 | -- | Body weight (yearling) QTL (66889); Udder swelling score QTL (106708); Interval to first estrus after calving QTL (30300) |
| 2 | 25 | 86178360 | 88823250 | PGAP1, ANKRD44, RFTN2, BOLL, PLCL1, SATB2, FBX036 | Milk fat yield QTL (122473) |
| 2 | 28 | 119194847 | 132528595 | ECE1, ALPL, SLC16A14, SP140, SP110, SP140L, CAB39, PSMD1, SPOCD1, FABP3, SNRNP40, LAPTM5, HSPG2, USP48, RAP1GAP | Fat thickness at the 12th rib QTL (126458); Tick resistance QTL (135875); Milk fat yield (daughter deviation) QTL (25782); Lignoceric acid content QTL (19771, 19709) |
| 3 | 36 | 101384522 | 105043063 | GPBP1L1, PRDX1, TESK2, ZSWIM5, PTCH2, KIF2C, C3H1orf228, RNF220, ERI3, SLC6A9, ST3GAL3, SLC2A1, PPCS, FOXJ3 | Body weight (weaning) QTL (24748); Birth index QTL (15168); Calf size QTL (15167, 15169); Calving ease QTL (15170) |
| 5 | 25 | 55540165 | 56247946 | ANKS1B, AVIL, TSFM, METTL21B, METTL1, CYP27B1, MARCH9, CDK4, AGAP2, OS9 | Inhibin level QTL (71509, 71314, 71315, 71316, 71317, 71358, 71359, 71319, 71407, 71320); Coat color QTL (37323, 37324, 37325) |
| 5 | 40 | 62920850 | 66830677 | ACTR6, NR1H4, ANO4, SLC5A8, UTP20, PARPBP, NUP37, GNPTAB | Fat thickness at the 12th rib QTL (20283); Calving ease QTL (126849); Stillbirth QTL (126850); Gestation length QTL (15409, 15410, 15411, 15412); Milk C14 index QTL (34847); Lean meat yield QTL (36911) |
| 5 | 14 | 103006312 | 104069660 | WC1-12, WC1.3, CLSTN3 | Shear force QTL (121704) |
| 6 | 19 | 24733464 | 26517604 | PPP3CA, EMCN, DNAJB14 | Clinical mastitis QTL (25244); Body weight (birth) QTL (67220, 67402, 66543); Body weight gain QTL (67403, 67639); Calving ease QTL (106434) |
| 6 | 16 | 73543373 | 75270817 | KIAA1211, AASDH, SRP72, NOA1, IGFBP7 | Curd firmness QTL (95977); Eye area pigmentation QTL (37389); Body weight (birth) QTL (66358); Body weight (weaning) QTL (67229); Body weight (yearling) QTL (67230); Body weight gain QTL (67231); Milk yield (EBV) QTL (16233); Milk fat yield QTL (16234); Milk fat percentage (EBV) QTL (16235); Milk protein percentage QTL (16236); Non-return rate (EBV) QTL (16237); Interval from first to last insemination QTL (16238); Milk protein yield (daughter deviation) QTL (26193, 26196); Milk fat yield (daughter deviation) QTL (25834); Milk yield (daughter deviation) QTL (25421); Milk kappa-casein percentage QTL (111104, 112361) |
| 7 | 9 | 4358132 | 4953801 | CRTC1, CRLF1, ELL, SSBP4, PGPEP1 | Body weight gain QTL (67930) |
| 7 | 56 | 94824183 | 100624993 | NR2F1, FAM172A, KIAA0825, SLF1, MCTP1, FAM81B, TTC37, PCSK1, ERAP1, LNPEP, LIX1 | Zinc content QTL (24065); Fat thickness at the 12th rib QTL (24649, 24650); Shear force QTL (20767, 31580, 37955); Iron content QTL (23257, 23258); Lean meat yield QTL (37087); Carcass weight QTL (122454); Tenderness score QTL (36406) |
| 8 | 45 | 46925080 | 55060853 | SMC5, TRPM3, ABHD17B, C8H9orf85, GDA, ZFAND5, TMC1, RFK, PRUNE2, GNA14, VPS13A, GNAQ, CEP78 | Average daily gain QTL (20937); Zinc content QTL (24066); Milk yield QTL (121783); Milk fat yield QTL (121784); Milk protein yield QTL (121785); Milk protein percentage QTL (121786) |
| 8 | 29 | 74227581 | 77959797 | DOCK5, CDCA2, PPP2R2A, PTK2B, GULO, B4GALT1, NFX1, UBE2R2, KIF24, DNAI1, CCL19 | Milk protein yield (daughter deviation) QTL (26264, 26268); Milk fat yield (daughter deviation) QTL (25878); Milk yield (daughter deviation) QTL (25466); Fat thickness at the 12th rib QTL (122442); Interval to first estrus after calving QTL (29993, 30322) |
| 10 | 12 | 28781938 | 29967808 | FMN1, AVEN, RYR3 | Docosapentaenoic acid content QTL (31774); Omega-3 unsaturated fatty acid content QTL (31780); Calving ease (maternal) QTL (44416); Daughter pregnancy rate QTL (44417); Stillbirth (maternal) QTL (44418); Foot angle QTL (44419); Feet and leg conformation QTL (44420); PTA type QTL (44421); Teat placement (front) QTL (44422); Udder attachment QTL (44423); Net merit QTL (44424); Length of productive life QTL (44425); Rear leg placement - rear view QTL (44426); Rear leg placement - side view QTL (44427); Udder height QTL (44428); Rump width QTL (44429); Calving ease QTL (44430); Somatic cell score QTL (44431); Stillbirth QTL (44432); Stature QTL (44433); Udder depth QTL (44434) |
| 10 | 52 | 30065944 | 39409071 | DPH6, C10H15orf41, MEIS2, RTF1, MGA, MAPKBP1, SPTBN5, PLA2G4E, VPS39, CAPN3, STARD9, CDAN1, UBR, SCG5, AQR | Residual feed intake QTL (23793); Iron content QTL (24060); Calving ease QTL (15185, 44566); Dry matter intake QTL (140483); Body depth QTL (44557, 44572, 44586, 44672); Dairy form QTL (44558, 44573, 44587, 44673); Daughter pregnancy rate QTL (44559); PTA type QTL (44560, 44575, 44589, 44675); Net merit QTL (44561); Length of productive life QTL (44562); Rear leg placement - rear view QTL (44563, 44578, 44592, 44677); Teat placement - rear QTL (44564, 44579, 44593); Udder height QTL (44565, 44580, 44594, 44678); Somatic cell score QTL (44567); Stillbirth QTL (44568); Stature QTL (44569, 44582, 44596, 44680); Teat length QTL (44570); Udder cleft QTL (44571, 44584, 44598); Conception rate QTL (107126, 107124); Feet and leg conformation QTL (44574, 44588, 44674); Teat placement - front QTL (44576, 44590); Udder attachment QTL (44577, 44591, 44676); Rump width QTL (44581, 44595, 44679); Strength QTL (44583, 44597, 44681); Udder depth QTL (44585, 44599); Shear force QTL (37956, 20778) |
| 10 | 60 | 42713194 | 49942490 | KLHDC2, NEMF, SOS2, CDKL1, MAP4K5, TRIM9, CSNK1G1, FAM96A, DAPK2, HERC1, CA12, APH1B, TLN2, VPS13C, FRMD6, GNG2, NID2, PTGDR, ZNF609, TRIP4, RORA, ICE2 | Dairy form QTL (44720); Daughter pregnancy rate QTL (44721); Net merit QTL (44722); Length of productive life QTL (44723); Milk protein percentage QTL (44724, 105566); Calving ease QTL (44725); Somatic cell score QTL (44726); Stillbirth QTL (44727); Milk glycosylated kappa-casein percentage QTL (116745, 111446); Milk fat yield (daughter deviation) QTL (25900); Tick resistance QTL (135843); Dry matter intake QTL (131016, 131002); Body weight (birth) QTL (68188); Body weight gain QTL (68135); Eicosapentaenoic acid content QTL (31775); Omega-3 unsaturated fatty acid content QTL (31781) |
| 10 | 81 | 50086640 | 59882200 | DMXL2, GLDN, TNFAIP8L3, SPPL2A, TRPM7, MYO1E, CCNB2, ADAM10, LIPC, ALDH1A2, POLR2M, MYZAP, CGNL1, TCF12, NEDD4, PRTG, RAB27A, UNC13C, WDR72, FAM214A, MYO5A, GNB5, MAPK6 | Tick resistance QTL (135792); Body weight (weaning) QTL (23796); Udder swelling score QTL (106594); Interval to first estrus after calving QTL (28678, 28652); Fat thickness at the 12th rib QTL (122444); Bovine respiratory disease susceptibility QTL (95662); Body weight (weaning) QTL (23795); Conception rate QTL (138570); C22:1 fatty acid content QTL (20512); Calving ease QTL (30516); Shear force QTL (106393) |
| 10 | 63 | 60004650 | 66740333 | SHC4, CEP152, FBN1, SLC12A1, MYEF2, SEMA6D, FERMT2, DDHD1, GABPB1, ATP8B4, FAM227B, GALK2 | Calving ease QTL (30516); Shear force QTL (106393); Calving index QTL (30512); Pregnancy rate QTL (65946); Milk zinc content QTL (70034); Conception rate QTL (107130); Body depth QTL (44827, 44850, 44863); Dairy form QTL (44828, 44851, 44864); PTA type QTL (44829, 44853, 44865); Udder attachment QTL (44830, 44855, 44867); Udder height QTL (44831, 44857, 44869); Rump width QTL (44832, 44858, 44870); Stature QTL (44833, 44859, 44872); Strength QTL (44834, 44860, 44873); Udder cleft QTL (44835, 44861, 44874); Udder depth QTL (44836, 44862, 44875); Feet and leg conformation (44852); Teat placement - front QTL (44854, 44866); Teat placement - rear QTL (44856, 44868); Body weight gain QTL (68136), Somatic cell score QTL (44871); Body weight (weaning) QTL (106643); Gestation length QTL (15473, 15474) |
| 10 | 3 | 98223275 | 98398966 | -- | Body weight (weaning) QTL (24729) |
| 10 | 14 | 100918254 | 101806982 | GALC, KCNK10, ZC3H14, EML5 | Longissimus muscle area QTL (138414) |
| 11 | 28 | 2254541 | 4099982 | ASTL, CIAO1, KANSL3, LMAN2L, CNNM3, SEMA4C, FAM178B | Milk glycosylated kappa-casein percentage QTL (116678) |
| 11 | 31 | 36863940 | 40183335 | ZAP70, TMEM131, VWA3B, CNGA3, COA5, MGAT4A, KIAA1211L, ACYP2, SPTBN1, EML6, RTN4, CCDC88A, PPP4R3B, CCDC85A | Milk riboflavin content QTL (64136); Body depth QTL (45332); Calving ease (maternal) QTL (45333); Foot angle QTL (45334); Feet and leg conformation QTL (45335); Milk fat percentage QTL (45336); PTA type QTL (45337); Udder attachment QTL (45338); Milk fat yield QTL (45339); Milk yield QTL (45340); Net merit QTL (45341); Milk protein percentage QTL (45342); Milk protein yield (45343); Rear leg placement - rear view QTL (45344); Udder height QTL (45345); Rump width QTL (45346); Calving ease QTL (45347); Stillbirth QTL (45348); Stature QTL (45349); Strength QTL (45350); Udder depth QTL (45351); Shear force QTL (36420, 36421); Udder swelling score (106598); Dry matter intake QTL (121918); Calving interval QTL (121653, 121654) |
| 13 | 25 | 61014137 | 63715266 | ANGPT4, TBC1D20, HM13, TTLL9, CCM2L, NOL4L, DNMT3B, SUN5, BPIFB4, SNTA1 | Teat length QTL (47334); Interval to first estrus after calving QTL (28680); Milk iron content QTL (70225); Scrotal circumference QTL (119789) |
| 14 | 10 | 4202927 | 4797465 | KCNK9, TRAPPC9, AGO2 | Milk fat percentage QTL (33087, 35538, 47482, 61978, 32849, 35540, 14973, 33665, 33581, 35542, 33227, 32960); Milk protein percentage QTL (35774, 113879, 47486, 35776, 113990); Milk protein yield QTL (35306, 35308, 14927); Milk yield QTL (35423, 35425, 14900); Milk casein percentage QTL (108945); Milk riboflavin content QTL (64384); Somatic cell score QTL (64598); Foot angle QTL (47480); Feet and leg conformation QTL (47481); Milk fat yield QTL (47483, 35359, 35360, 31117, 35362); Net merit QTL (47484); Length of productive life QTL (47485); Rear leg placement - rear view QTL (47487); Rear leg placement - side view QTL (47488); Calving ease QTL (47489); Stillbirth QTL (47490); Stature QTL (47491); Strength QTL (47492); Milk oleic acid percentage QTL (32556) |
| 14 | 18 | 33747933 | 35216192 | C14H8orf34, PREX2, CPA6 | Udder swelling score QTL (106736); Age at first calving QTL (140104); Scrotal circumference QTL (138445) |
| 16 | 25 | 2020646 | 3805373 | PIK3C2B, NFASC, CNTN2, DSTYK, TMCC2, MFSD4A, NUCKS1, PM20D1, CTSE, SRGAP2 | Body depth QTL (48079); Calving ease (maternal) QTL (48080); Dairy form QTL (48081); Daughter pregnancy rate QTL (48082); PTA type QTL (48083); Length of productive life QTL (48084); Calving ease QTL (48085); Teat length QTL (48086); Udder cleft QTL (48087); Milk myristic acid percentage QTL (56623); Body weight gain QTL (68840) |
| 16 | 11 | 5715970 | 7838108 | KCNT2 | Milk fat percentage QTL (34602) |
| 16 | 73 | 25709923 | 26613598 | -- | -- |
| 20 | 40 | 21402231 | 22154025 | GPBP1 | Body weight gain QTL (69057) |
| 20 | 36 | 27105033 | 30747366 | HCN1, EMB, PARP8 | Calving ease QTL (30553); Average daily gain QTL (131122); Length of productive life QTL (123122, 123133); Milk protein percentage QTL (105307, 105349, 105182, 105395, 105073); Milk protein percentage (daughter deviation) QTL (26866, 26984); Milk yield (daughter deviation) QTL (25661); Body weight QTL (65983) |
| 20 | 15 | 45232289 | 47209012 | CDH9 | -- |
| 21 | 6 | 6388853 | 6900116 | CERS3, ADAMTS17 | Body depth QTL (50966); Calving ease (maternal) QTL (50967); Daughter pregnancy rate QTL (50968); Stillbirth (maternal) QTL (50969); Foot angle QTL (50970); Feet and leg conformation QTL (50971); PTA type QTL (50972); Teat placement - front QTL (50973); Udder attachment QTL (50974); Net merit QTL (50975); Length of productive life QTL (50976); Milk protein percentage QTL (50977); Udder height QTL (50978); Rump width QTL (50979); Calving ease QTL (50980); Stature QTL (50981); Strength QTL (50982); Udder depth QTL (50983) |
| 21 | 27 | 7068248 | 9209367 | MEF2A, LRRC28, TTC23, SYNM, IGF1R, PGPEP1L | Age at puberty QTL (21135, 21136); Sire conception rate QTL (124003); Longissimus muscle area QTL (22856); Dry matter intake QTL (23894); Body weight QTL (22618); Interval from first to last insemination QTL (138530); Inseminations per conception QTL (138531) |
| 21 | 9 | 63970044 | 65102247 | -- | -- |
| 22 | 18 | 21888915 | 23828165 | TPR1, SUMF1, CNTN4 | M. paratuberculosis susceptibility QTL (127097); Body weight (birth) QTL (23910) |
| 22 | 1 | 50772465 | 50772465 | -- | -- |
| 24 | 6 | 33291018 | 33669073 | LAMA3, ANKRD29, NPC1, TMEM241 | Milk protein yield QTL (123993) |
| 29 | 7 | 50202589 | 50336324 | TSPAN32 | Average daily gain QTL (102011, 102012); Growth index QTL (102013) |
Discussion
Principal component analyses
The PCA is a useful tool that allows farmers to identify genetically different individuals within their herd based on genotypes. The graphical representation of individuals in fact represent an immediate easy to interpret tool that does not require any specific skill by farmers except the concept that closed points are more similar and distant ones are different. Well-differentiated females can be identified in this herd, mostly due to sire, although a group appearing genetically similar exists belonging to a variety of sires. Sires 1 and 5 are responsible for the out-groups we see, with their derivations being Canada and Italy, respectively. Therefore, as proposed by the farmer involved in this study, this visual is beneficial to determine the a-posteriori analysis of his mating strategies and whether the sires he had chosen are helping to his attempts to limit the reduction in genetic variability of the herd. The PCA is also beneficial in sire selection, as the farmer can ascertain if sires deriving from specific countries have an impact on the genetic distribution in the population. Likewise, it can be visualized how genomic values, in this case GPFT, coincide with different individuals. GPFT of similar value did not cluster, indicating that both sire and sire origin did not impact GPFT in the females of this herd. Sire 1 contributed to individuals with GPFT values ranging from 1,500 to 3,000 and sire 5 to animals with values from 1,000 to 2,500, for example.
Inbreeding coefficients
This result here obtained are concordant with another study performed in Italian Holstein, where the average F PED was 0.044 and the F ROH >1 Mb was 0.1165. Across all 44 animals, the correlation between F PED and F ROH was significantly different from 0 with r equal to 0.453 (P<0.01). Intense and accurate artificial selection practiced over many years has resulted in high rates of genetic gain; however, the high rates of gain have been accompanied by large increases in inbreeding17. Additionally, as population sizes decrease, the probability of mating among relatives increases, especially in small herds or local breeds13. Producers have largely depended on pedigrees to estimate the coefficient of inbreeding within the herd, but this number can only increase with each generation. Pedigree relatedness gives an expected, not actual, proportion of genomic identity by descent among individuals and it is anticipated that genotype-based estimates provide greater accuracy on relatedness18. In practice, pedigree information is difficult to obtain, potentially unreliable, and rarely assessed for inbreeding arising from common ancestors19. When the pedigree-based coefficients of this herd was examined, it was possible that individuals to be up to a tenth inbred. But, in the absence of pedigree data, the extent of a genome under ROH may be used to infer aspects of recent population history, even from relatively few samples3,9. McQuillan et al.9 revealed that F ROH correlates strongly with the inbreeding coefficient estimated from pedigree (r= 0.86), and it has been found that F ROH values are preferable to F PED, as they are thought to be a more realistic reflection of inbreeding level15,19. However, it is important to note that the setting of the parameters used to derive ROH is crucial to account for the effects of SNP density correctly20. In this dataset, it is visible a linear relationship between F ROH and F PED. The Pearson’s correlation coefficient provides a positive correlation at r= 0.453. The correlation in this work is in concordance with other studies using cattle that were genotyped at similar density15,20, yet lower than others that were genotyped at medium to high density3,5. It is possible then that the correlation coefficient may be affected partially by the small number of animals in this study as such as for the use of a low-density SNP chip. Additionally, the F ROH was larger than the mean F PED in 35 animals, implying that pedigree based inbreeding coefficients could be underestimated.
The overall low F IS values are indicative of a low level of inbreeding in the population and of a relatively high number of individuals in a heterozygous state. This may be related to the result of the specific mating strategy implemented by the University herd manager co-author of this work, as he stated his goal was to increase genetic variability and preserve low inbreeding. Therefore, this genomic information is providing unique feedback as to the successful efforts in maintaining inbreeding within this population. Dairy breeds especially have been under more intensive selection and may be related to the recent increase of consanguineous mating resulting from small number of high genetic merit sires used for artificial insemination5. Because of this, it is important to understand the inbreeding occurring at a genomic level, as well as to determine whether or not the genetic variability within one’s herd is being well maintained.
Runs of homozygosity and associated genes
The most commonly observed QTL within these regions reported in Table 2 included milk protein percentage, milk fat, calving ease, PTA type, and stature. After aligning the SNP to the BTAU 5.0.1 reference, 260 annotated genes were identified to be consistent with the top 5% of SNP, while 37 genes remained in the top 1%. Genes in the top 1% resided on chromosomes 5 and 10; no annotated genes were identified with the SNP on BTA 16. STRING subsequently identified networks amongst these genes, with 142 genes identified to be involved in some kind of network (Figure 5). These genes also underwent gene ontology in PANTHER. Genes within the top 5% corresponded with 12 biological processes (Figure 6) and 70 pathways (data not shown). The pathway that included the greatest number of genes (n= 12) was the gonadotropin-releasing hormone receptor pathway, while other pathways sharing genes were the 5HT2 type receptor mediated signaling pathway, endothelin signaling pathway, inflammation mediated by chemokine and cytokine signaling pathway, integrin signaling pathway, and Wingless-related integration site (Wnt) signaling pathway. The respective biological process most commonly shared among genes (n= 116) was “cellular processes,” specifically cell communication, while another large portion (n= 79) has a role in metabolic processes (Figure 6).

Figure 5 Network of genes corresponding with SNP in the top 5% of incidence of common runs obtained by STRING online database
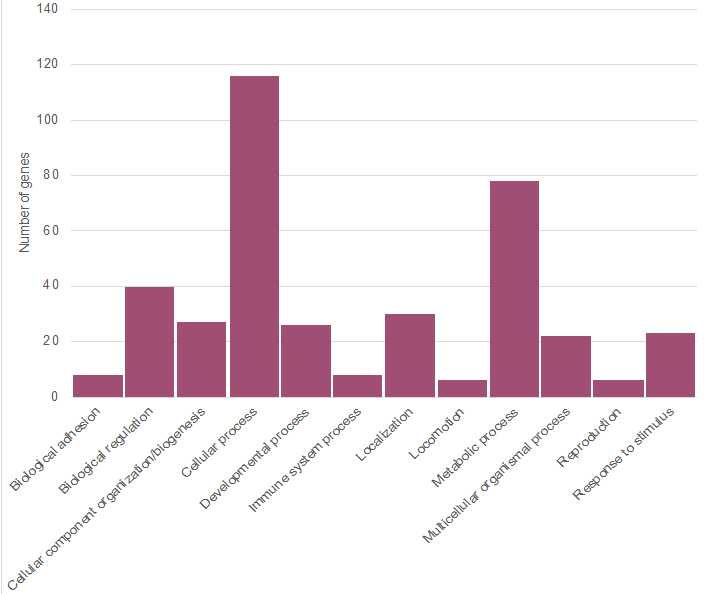
Figure 6 Biological processes of genes corresponding with SNP in the top 5% of incidence of common runs
The practices of intense selection of sires, artificial insemination, and embryo transfer have been featured heavily in some breeds, reducing effective population sizes, genetic diversity and affecting levels of homozygosity3. The deleterious effects associated with boosted homozygosity arising from inbreeding are predisposed to reduce the genetic gains, implicating in a clear loss of genetic variability15. ROH give insight to this variability, as they are continuous homozygous segments that are common in individuals and populations. The ability of these segments to give a glimpse into a population’s genetic events makes them a useful tool in providing information about the evolution of the population over time, therefore enabling producers to maintain diversity and fitness within their livestock breed21. Furthermore, ROH provide useful information about the genetic relatedness among individuals, helping to minimize the inbreeding rate and also helping to expose deleterious variants in the genome21. Long ROH arise as a result of recent inbreeding, while shorter ROH can indicate more distant ancestral effects such as breed founder effects3. In point of fact, the presence of segments longer than 10 Mb is traceable to inbreeding from recent common ancestors that occurred only five generations ago22, and 66 % of the animals comprised in this study presented at least one ROH extending over 10 Mb. This signifies that the majority of individuals in this population derived from recent common ancestors and is the product of recent inbreeding, consistent with what we see from the inbreeding coefficients.
ROH were also identified as a means to provide insight to the conserved genomic regions within the herd. ROH less than 5 Mb were recently characterized as being short23. These shorter ROH can be related to a more distant ancestral positive selection effect due to recombination events from repeated meiosis breaking long chromosomes into segments and therefore reducing their size13,24. Lower SNP density, such as that used in this study, tends to inflate ROH3. Given that we see primarily short ROH in this herd (average ROH=4.66 Mb), even with the potential inflation, it is possibly to say with confidently that the level of inbreeding within this population has been well maintained, is low, and is recent based upon the presence of long ROH in the majority of individuals yet the overall short ROH throughout the herd. This result identifies with a separate study that established that the Italian Holstein show higher number of ROH segments related to ancient consanguinity13.
Genes within the extended homozygous regions corresponding to SNPs in the top 5% of common runs were found to be largely involved in survival processes. This indicates that basic biological processes, such as cell communication and metabolism, have been maintained in selection efforts. QTL within these regions, however, show that these genomic regions coincide with beneficial production facets such as milk protein percentage, milk fat, and calving ease, although these may be specific to the Holstein-Friesian breed due to stringent selection efforts over time. Additionally, genes in the top 1% (BTA 5, 10, and 16) also correlated with survival and developmental processes. Such processes included cell proliferation (GNG2, ADAM10, ALDH1A2), metabolic function (SCG5, PTGDR, RORA, MYO1E, LIPC, ALDH1A2, MYO5A, GALK2), organ development (ANKS1B, MYO1E, CCNB2, ALDH1A2, PRTG, MAPK6), and immunity (NEDD4). No genes were identified on BTA 16 due to poor annotation, but several genes on BTA 5 and 10 have been well characterized in different phenotypes in cattle. PARP1 binding protein (PARPBP) on BTA 5 has been associated with fat percentage in the third lactation stage in Jersey cattle, while ankyrin repeat and sterile alpha motif domain containing 1B (ANKS1B), also on BTA 5, has been associated with bovine spongiform encephalopathy (BSE) incidence in cattle families25,26. Meanwhile, genes on BTA 10 were largely found to be involved in fat metabolism and feed intake. Secretogranin V (SCG5) and G protein subunit gamma 2 (GNG2), for example, have been linked to the regulation of hormone metabolic processes involved in feed intake in Holstein27,28, while transcription factor 12 (TCF12) was found to be associated with lipid and organoleptic traits in European Bos taurus breeds29. GNG2 was also found to be in a region associated with susceptibility to Mycobacterium avium ssp. Paratuberculosis (Map) infection30. Lastly, FERM domain containing 6 (FRMD6) was identified to be in a region of positive selection in the N’Dama breed31. The regions on BTA 10 and 16 harboring the top 1% of SNP were also concordant with other studies. Studies involving Holstein were especially similar, with positive selection occurring on BTA 10 at 50-60 Mb and a putative QTL identified at 34.7-56.9 on BTA 10 for somatic cell count and non-return rate at 90 days (paternal effect)20,32,33. Another study identified a high proportion of ROH on BTA 16, while a separate found selection at regions 24.7-26.7 and 26.5-28.5 on BTA 163,20.
Conclusions and implications
Using a population of 44 Italian Holstein-Friesian cattle from the University experimental farm and low-density SNP panels for genotyping, we estimated several genetic variability aspects within the herd population including inbreeding coefficients and runs of homozygosity. The approach was successful to provide a tool to monitor the efficacy of the mating strategies operated in the farm population and to provide insights for the inbreeding management. SNP genotypes can effectively provide inbreeding coefficients in the absence of a pedigree, while runs of homozygosity can further provide information on genomic regions under positive selection and thus indicate the evolving nature of the population. If farmers genotype their young females more extensively, they are able to construct a database with the corresponding information and are therefore enabled to make selection and management decisions as a result. It is to be noticed that the management of the inbreeding is extremely important to reduce the occurrence of mendelian recessive disease and that the genomic approach is providing an innovative and accurate possibility. The farmers are usually genotyping females with low density SNP chip that is shown to be a valuable tool to be used for genomic management purposes. The diffusion of this approach to farmers may lead to a routine genotyping of young females thus providing information for a wide impact of the genomic management of inbreeding.

