











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkGenetic improvement depends on genetic variation, selection intensity, generation interval, and accuracy of estimated breeding values (EBV). In the genetic evaluation of animals, it is important to maximize EBV accuracy. An increase in EBV accuracy for selection of candidate animals will spur genetic progress. Among other advantages, the use of genomic selection allows an increase in the accuracy of genetic values1, especially at a young age2. Three technological breakthroughs have boosted wide-spread DNA information use in animal breeding3: the development of genomic selection technology, the discovery of massive numbers of genetic markers (SNPs), and high throughput cost-effective genotyping technology. Although the advantages of genomic selection have been observed most notoriously in dairy cattle4, in general, the use of genomic selection can be expected to yield improvements in genetic progress of up to 10 % in any species4.
In genomic evaluation, response variables can be individual phenotypes, repeated observations, records on close family members such as progeny, EBVs or their deregressed counterparts from genetic evaluations5,6. According to these authors, using deregressed EBV (DEBV), an accuracy of up to 2.76 times higher than with records of a single individual can be obtained. With average daily gain and feed conversion ratio of swine data, obtained accuracies were 18 to 39 % higher, depending on the trait evaluated, when DEBVs were used as response variables instead of EBVs7. These authors concluded that DEBV is the preferred response variable, whereas the choice of statistical method was less critical when they analyzed purebred swine data. The increase of 18 to 39 % in reliability is worthwhile, since the reliabilities of the genomic breeding values directly affect the returns from genomic selection7.
Deregressed EBVs, with the parent average removed, produce more exact predicted genomic values (GV) for two reasons5. First, DEBVs, when used as the response variable, result in fewer double counts than when EBVs are used because the DEBVs exclude information from the individual’s ancestors. If both the offspring and its parents are genotyped, the degree of double count decreases when DEBVs are used as the response variable. Second, when using EBVs as the response variable, the degree of double count in the GVs decreases, particularly when the reliabilities of the genetic values are low.
However, DEBVs are not always the best choice for use as the response variable in genomic evaluation. Simulated dairy cattle6 and jumping horse8 data were used to compare EBVs and DEBVs as response variables. Both groups of authors found only slight advantages to using DEBVs, instead of conventional EBVs, as response variables. The objectives of this study were to compare the accuracy of genomic values and to estimate the genetic correlation between true genetic values and genomic values obtained using predicted breeding values (EBV) and deregressed EBV (DEBV) as response variables for four training populations and four evaluation generations.
The methodology for simulating the training (PEn) and evaluation (PEv) populations used in this study was described previously9. Briefly, two populations using the QMSim program10 were simulated. The first, to obtain linkage disequilibrium, had 800 individuals as the effective population size and 100 generations. The second population, where PEn and PEv originated, had 14 discrete generations, each of which was generated randomly using 20 males and 200 females, a panel of 53,010 SNPs (each evenly separated by 100 centiMorgans) randomly placed in 30 chromosomes, and 540 QTLs with effects coming from a gamma distribution11. Both SNPs and QTLs were regarded as biallelic with random starting frequencies. Genotypes and phenotypes of 6,400 individuals were simulated; the heritability used was 0.4 and only additive effects were considered. Genotypic and phenotypic information was generated using the QMSim program10. The four PEn comprised generations 10 (n= 1,000); 9 and 10 (n= 1,400); 8 to 10 (n= 1,800); and 7 to 10 (n= 2,200); as well as their phenotypes and the corresponding EBVs and DEBVs. The four PEv comprised generations 11 to 14.
In a first step, the EBVs were predicted with a single-trait animal model including the random effect of animal, the fixed effects of sex of the individual, and generation. The ASREML program12 was used at this stage. The DEBVs were then obtained following methodology of Garrick et al5. Weight (wi) for the ith animal was obtained using the following equation5:

Where c is the lack of fit of the prediction equation, or the genetic part not explained by the markers5; the value assumed was c=0.1; heritability of the trait, h2, was assumed to be 0.4; and r2 was the reliability of the DEVGs for the ith animal.
Deregression of EBVs adjusts for ancestral information, it removes shrinkage present in EBV, and by taking parental contribution into account, DEBVs can be regarded as equivalent to the information provided by the records of each sire and its progeny13.
In a second step, the predicted genetic values (EBVs) obtained using ASREML and their corresponding DEBVs were used as response variables to predict the GVs. A weighted genomic analysis was carried out using the BayesCπ function of the Gen-Sel program14. A 41,000-round long chain was used. The last 1,000 samples were used to obtain the a posteriori mean estimates of marker effects and variances. The first 40,000 iterations were regarded as the burn-in period; π was fixed at 0.95. The genomic analysis used animals of generations 7 to 10 to obtain the prediction equations. The evaluation populations were generations 11 to 14. The Bioinformatics to Implement Genomic Selection (BIGS) platform (http://bigs.ansci.iastate.edu/) platform was used for the analysis.
The genomic values and their corresponding accuracies were obtained by summing all the SNP effects, using the following equation:
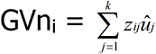
Where GVni is the genomic value for the ith individual; zij is the genotype of the jth marker on the ith individual, and ûj is the a posteriori mean of SNP effect for the jth marker.
Accuracies (R2) of GVs were obtained as the square of the correlation between GVs and the true genetic values6,13,15. Criteria for comparing the two alternatives of analysis were R2 and GV prediction error variance (PEV). Additionally, as another criterion for comparing the two response variables studied, the genetic correlation estimates was used between the true genetic values and the predicted GVs from the two alternatives of genomic analysis6. These estimates were obtained using ASREML12.
An important aspect in genetic improvement is the response to selection, and this depends on selection accuracy16. Table 1 shows the means and corresponding standard deviation for R2 and PEV of GVs obtained from the different combinations of PEn and PEv when the response variable was DEBV. The highest R2, 0.77 ± 0.01, was observed for the largest training population (generations 7 to 10, n= 2,200 individuals) and 11 was the generation under evaluation. In contrast, the lowest mean for R2, 0.28 ± 0.06, was observed for the combination of the smallest training population and the farthest evaluation population being evaluated, generation 14. These results are within the range of R2 values reported by Hassani et al17, who found 0.49 (±1 SNP) to 0.75 (±100 SNPs) using whole-genome training for single QTL with a 50 K SNP panel and BayesC0.
Table 1 Mean ± standard deviation for accuracy (R2) and prediction error variance (PEV) of genomic values obtained using deregressed predicted genetic values as response variables, four training populations, and four generations of evaluation
| Training population | ||||
|---|---|---|---|---|
| Evaluation generation | 10 | 9 and 10 | 8 to 10 | 7 to 10 |
| R2 | ||||
| 11 | 0.52±0.04 | 0.67±0.03 | 0.73±0.03 | 0.77±0.01 |
| 12 | 0.39±0.04 | 0.55±0.03 | 0.63±0.03 | 0.68±0.03 |
| 13 | 0.32±0.05 | 0.49±0.04 | 0.58±0.03 | 0.64±0.03 |
| 14 | 0.28±0.06 | 0.45±0.04 | 0.54±0.04 | 0.60±0.03 |
| PEV | ||||
| 11 | 0.05±0.003 | 0.05±0.003 | 0.04±0.003 | 0.04±0.003 |
| 12 | 0.06±0.004 | 0.06±0.004 | 0.06±0.004 | 0.05±0.004 |
| 13 | 0.07±0.005 | 0.07±0.005 | 0.06±0.005 | 0.06±0.005 |
| 14 | 0.07±0.006 | 0.08±0.006 | 0.07±0.006 | 0.07±0.005 |
Two clear trends can be observed for R2 in Table 1. First, as PEn and PEv moved farther apart, R2 decreased. Second, as the size of PEn decreased, R2 became smaller. These results are similar to those reported by other research groups18-21, who concluded that the closer the relationship between individuals in PEn and those in PEv, the higher the R2 of GVs. Similarly, using both simulated and real sheep data, Genomic Best Linear Unbiased Prediction was compared with two pedigree based methods22. It was found that both empirical and estimated accuracy of GVs were different for several degrees of relationship. These authors concluded that R2 of GVs is proportional to the genetic relationship of animals under selection to the reference population. The increase in R2 of GVs when PEn and PEv are closely related, can be explained by more precise genomic relationships, improving in this way the connectedness between these populations and more distant populations. Accordingly, another research group23 concluded that accuracy of GVs deteriorated as the relationship between animals in the PEn and those under selection decreased. One implication of this is that PEn has to be regularly updated to keep the marker effect estimates in sync with new generations of the breeding population2.
On the other hand, as expected, the trend for R2 held true for PEV, but in the opposite direction. The greater the population size and the closer relationship between PEn and PEv, the lower PEV. Pszczola et al15 mentioned that PEV can be calculated as the connectedness between the reference population and the animals under evaluation. This may explain the increase in PEV as PEn and PEv became farther apart. Greater connectedness reduces bias, and thus genetic evaluation improves24. The observed trend for PEV held true for both EBV and DEBV response variables.
Table 2 shows the means and their corresponding standard deviations for R2 and PEV for the combinations of PEn and PEv when the response variable was EBV. In general, R2 values were only slightly lower than those observed when DEBVs were used as response variables. The trends observed for the decrease in R2 and the increase in PEV when DEBVs were response variables, as size of PEn diminished and the distance between PEn and PEn augmented, held true for EBVs used as response variables. These results are similar to those obtained by other authors25,26,27, who found that size of PEn affected R2 of GVs. The results of the present study and those obtained by other groups of researchers agree with what could be theoretically expected1,28. These authors developed predictive equations for accuracy of predicted genomic values, which depend on size of PEn, effective population size of the breed, heritability of the trait, and length of genome.
Table 2 Mean ± standard deviation for accuracy (R2) and prediction error variance (PEV) of genomic values obtained using predicted genetic values as response variables, four training populations, and four generations of evaluation
| Training population | ||||
|---|---|---|---|---|
| Evaluation generation | 10 | 9 and 10 | 8 to 10 | 7 to 10 |
| R2 | ||||
| 11 | 0.48±0.04 | 0.65±0.03 | 0.71±0.02 | 0.76±0.02 |
| 12 | 0.38±0.05 | 0.55±0.03 | 0.62±0.03 | 0.68±0.02 |
| 13 | 0.32±0.05 | 0.49±0.03 | 0.58±0.03 | 0.64±0.03 |
| 14 | 0.28±0.06 | 0.45±0.04 | 0.54±0.04 | 0.60±0.03 |
| PEV | ||||
| 11 | 0.05±0.003 | 0.05±0.003 | 0.04±0.003 | 0.04±0.003 |
| 12 | 0.05±0.004 | 0.06±0.004 | 0.06±0.004 | 0.05±0.004 |
| 13 | 0.06±0.004 | 0.07±0.005 | 0.06±0.004 | 0.06±0.004 |
| 14 | 0.06±0.005 | 0.07±0.006 | 0.07±0.005 | 0.06±0.005 |
The results of a study with two multi-breed beef cattle populations and Angus and Hereford purebred populations13 used to obtain the GVs and corresponding R2 for six growth and carcass traits showed that accuracies were lower for prediction equations trained in a single breed. These results were attributed to the smaller number of records derived from a single breed in the training populations. The R2 range was 0.01 ± 0.10 to 0.65 ± 0.07, although the authors also reported a negative estimate, -0.10 ± 0.15.
The results of this work, regardless of whether DEBV or EBV were used as response variables, are similar to those obtained by Saatchi et al23. These authors evaluated different training populations of Hereford cattle; accuracy estimates ranged from 0.15 to 0.52, with 0.30 on average when trained on old animals and validated on young animal populations. The results obtained in our study may be explained by the fact that genomic prediction on closely related individuals is based on relationship; genomic relationships are more accurate when the relationships between PEn and PEv populations are close3. On the other hand, prediction on distant individuals requires DL between QTL and markers29.
The R2 results are lower than those reported by Pszczola et al30. These authors found that the inclusion of animals with predicted genotypes in the reference population did not significantly increase accuracies of GVs for juvenile animals. They attributed the lack of significance to the low accuracy of predicted genotypes and concluded that inclusion of non-genotyped animals is expected to enhance genomic selection accuracy only when the unknown genotypes can be predicted with high accuracy. The results obtained by these authors varied from 0.57 to 0.96, from 0.48 to 0.88, and from 0.33 to 0.72 for heritabilities of 0.30, 0.05, and 0.01, respectively, under different sizes of the reference population, and different numbers of animals with known or predicted genotypes.
The small difference in GV accuracy that we obtained in our study when EBVs or DEBVs were the response variables agree with reports by other researchers. However, these results are opposite to those observed by Ostersen et al7, who found 18 to 39 % higher accuracies for feed conversion ratio and daily gain when they used DEBVs instead of EBVs as response variables. The estimation methodologies they used were GBLUP, Bayesian Lasso, and MIXTURE, where the marker effects are assumed to follow a normal distribution, double exponential, and a mixture of two normal distributions, respectively. The three alternatives of analysis yielded similar reliabilities of the GVs for the two traits analyzed.
Contrary to our results, Ricard et al8 did not find substantial advantages to genomic values obtained using deregressed EBVs as response variables or the GBLUP and BayesC alternatives of analysis compared with conventional BLUP predictions. They followed a specific deregression procedure that included not only the individual’s own performance, but also the performance of several relatives (not just offspring), in addition to the genotyped sample. This regression procedure was easy to implement from EBVs, reliabilities, and pedigrees. Unfortunately, accuracy of genomic evaluation, measured by cross validation in several validation samples, was not enough to suggest its use in current breeding plans for the jumping horse population studied. However, the authors mention that this conclusion is related only to accuracy, and the potential benefits of a higher selection intensity, reduced generation intervals, and low inbreeding in the long run should be considered when genomic selection in horses is planned. In dairy cattle similar results were reported6. The authors compared two response variables, EBVs and daughter yield deviations (DYD) on simulated dairy data under eight scenarios of heritability, number of daughters per sire, and number of genotyped sires. They found that DYDs yielded slightly lower reliabilities than EBVs. The average differences in GV accuracy of between EBVs and DYDs were 0.009 for h2= 0.30, and 0.035 when h2= 0.05.
Table 3 presents the genetic correlation estimates between true genetic values and the GVs r(TBV,GV) obtained using DEBVs and EBVs as response variables. A slight advantage of using DEBVs, range 0.43 to 0.53, instead of EBVs, range 0.41 to 0.51, held constant throughout all training population sizes. Also, genetic correlation estimates decreased as PEn and PEv separated. The r(TBV,GV) estimates of the present study are higher than those observed by Alarcón-Zúñiga et al9, range 0.29 to 0.40, using the same dataset but different models for the genomic analysis. Genetic correlation estimates between direct genomic values and phenotypes from k-fold validation in Red Angus, Angus, Hereford, Simmental and Limousin ranged from 0.32 to 0.85 for birth weight, weaning weight, milk yield, rib eye muscle area, marbling, direct calving ease, and maternal calving ease21,31. Similarly, genetic correlation estimates between true genetic values and GVs for marbling, using data sets with different proportions of available information, ranged from 0.256 to 0.859 32. Guo et al6 found genetic correlation estimates between GVs and conventional parent average ranging from 0.457 to 0.688 using three statistical models and eight combinations of heritability and number of daughters per sire.
Table 3 Genetic correlation estimates between true genetic values and genomic values obtained using deregressed predicted genetic values (DEBV) or predicted genetic values (EBV) as response variables, with four training populations
| Response variable | ||
|---|---|---|
| Training population | DEBV | EBV |
| 10 | 0.53 | 0.51 |
| 9 y 10 | 0.51 | 0.50 |
| 8 a 10 | 0.48 | 0.47 |
| 7 a 10 | 0.43 | 0.41 |
Some limitations of our work are that a distance between training and evaluation populations needs to be more specific, and size and number of generations in the training population also need to be better determined. Moreover, since our study used simulated information, it does not entirely correspond to real production system conditions.
The advantage of using deregressed predicted genetic values as the response variable, instead of conventional predicted genetic values, was very slight with any combination of training population size and evaluation generation. Regardless of the response variable used, predicted genetic value or deregressed predicted genetic value, larger training population were associated with higher genomic values accuracy.
Prediction error variance was low and similar with any combination of training population size and evaluation generation, regardless of the response variable used. The genetic correlation estimates between true genetic values and genomic values obtained using DEBV as the response variable were slightly higher than those between true genetic values and genomic values obtained using EBV as the response variable.

