










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista mexicana de ciencias pecuarias
On-line version ISSN 2448-6698Print version ISSN 2007-1124
Rev. mex. de cienc. pecuarias vol.7 n.3 Mérida Jul./Sep. 2016
Articles
Isolation of microRNAs from Bouteloua gracilis chlorophyllous cells and their in silico characterization
a Facultad de Zootecnia y Ecología, Universidad Autónoma de Chihuahua. Periférico R. Almada Km. 1 C.P. 31453, Chihuahua, Chihuahua, México. Tel (614) 434-0303. Correo electrónico: evegonzal@uach.mx. Correspondencia al segundo autor.
b Programa de Posgrado, Instituto Tecnológico de Roque. Celaya, Guanajuato, México.
c Facultad de Ciencias Químicas, Universidad Autónoma de Chihuahua. Chihuahua, México.
Bouteloua gracilis is a grass native to Mexico, it is used as forage source for livestock because of its high nutritional value. It has high tolerance to osmotic stress and therefore can live in arid zones; however, regulation mechanisms in gene expression that confer these characteristics have not been reported. There is a class of small RNAs (sRNAs) called microRNAs (miRNAs), which regulate gene expression. They are complementary and act by binding to messenger RNAs (mRNAs to inhibit translation or by degrading them. In this work, the isolation of sRNAs from B. gracilis through cloning and sequencing of concatemers is reported. In silico analysis of the sequences obtained allowed to identify conserved sequences in Populus trichocarpa, Brachypodium distachyon, Oryza sativa, Sorghum bicolor, Zea mays, Malus domestica, and Linum usitatissimum. Furthermore, the secondary structure of the miRNA precursor (pre-miRNA) was predicted from two sequences isolated from In silico analysis using Glycine max, Zea mays, Sorghum bicolor, and Oryza sativa as reference genomes. Finally, six target mRNAs were identified for one of the miRNAs obtained. Identification of miRNAs in Bouteloua gracilis will help to understand how these molecules regulate gene expression and in the future will allow for the study at molecular level, providing insight on how this grass responds to environmental stress.
Key Words: Blue gramma; miRNAs; Genetic regulation; Bioinformatics analysis
Bouteloua gracilis es un pasto nativo del norte de México, que es utilizado como fuente de forraje para el ganado por su alto contenido nutritivo; tiene elevada tolerancia al estrés osmótico que le permite vivir en climas secos; sin embargo, los mecanismos moleculares que le confieren esta tolerancia aún no se han reportado. Existe una clase de RNAs pequeños (sRNAs) llamados microRNAs (miRNAs), que se unen por complementariedad a RNAs mensajeros blanco, etiquetándolos para su degradación o supresión de la traducción. En este trabajo se reporta el aislamiento de sRNAs de B. gracilis, a través de la clonación de concatámeros y su secuenciación. El análisis in silico de la secuencias, permitió la identificación de miRNAs conservados en B. gracilis y reportados en Populus trichocarpa, Brachypodium distachyon, Oryza sativa, Sorghum bicolor, Zea mays, Malus domestica y Linum usitatissimum. Además, se predijo la estructura secundaria de los precursores de dos miRNAs (pre -miRNAs), usando como referencia los genomas de Glycine max, Zea mays, Sorghum bicolor y Oryza sativa. Finalmente, se identificaron seis mRNAs blanco para uno de ellos. La identificación de miRNAs en Bouteloua gracilis, ayudará a comprender como estas moléculas regulan la expresión genética en esta especie, y sus relaciones con los mecanismos de respuesta a estrés ambiental.
Palabras Clave: Navajita azul; miRNAs; Regulación genética; Análisis bioinformático
Introduction
Sequences of miRNAs are single stranded RNA in 18 to 25 nucleotides in length; on plants, their biogenesis starts when miRNAs genes are transcribed by RNA polymerase II, generating primary transcripts with lengths of 200 to 300 nucleotides referred to as pri-miRNAs, which are transcripts containing a short internal stem-loop structure near the ribonuclease III (DCL), generating a pre- miRNA duplex. This duplex is transported from the nucleus to the cytoplasm where it joins the complex Argonauta (AGO) forming the RNA- induced silencing complex (RISC); finally, the complex binds to messenger RNAs complementary to the target, inducing degradation or suppression of translation, the miRNAs are considered silencing molecules of gene expression1.
In plants, these molecules are involved in the regulation of different physiological stages, as the change of the vegetative to the floral development and growth of leaves, stems, roots and reproductive organs. It has also been found that miRNAs regulate plant response to various abiotic and biotic stress such as lack of nutrients, salinity, drought, oxidative stress, cold, UV radiation and the presence of virus2,3. In silico studies aimed at orthologous plant miRNAs have shown that miRNAs are conserved between species; however, there are also species-specific miRNAs4. Mature miRNAs in plants are preserved to a greater extent than pre-miRNAs2,5. Most miRNAs that are conserved in different species homologous genes regulate5, this feature allows to analyze and predict potential targets messenger RNAs of miRNAs in species that have its genome sequenced. Predicting target mRNA is usually essential to identify the role of miRNA, this is done by bioinformatics analysis. The main criterion used is the detection of complementary sequences between the miRNA and its target6. This is possible because the complementarity between them is perfect or nearly perfect, allowing quick identification and reliable target mRNAs2,7.
Bouteloua gracilis (Willd. Ex Kunth) Lag. ex Griffiths is a perennial grass belonging to the Poaceae family with C4 metabolism are of thin stem that grow in forests, grasslands and thickets8,9. They are found in semi-arid regions of the United States and Mexico10. This grass is of great economic importance, because it is a source of fodder for feeding cattle and native wildlife, for its high protein content and high digestibility, and its ability to adapt to adverse weather conditions such as drought and cold11,12. Due to the above features, this grass has been subject to various ecological and physiological investigations. However, to date there is no information about the miRNAs expressed in B. gracilis and therefore its relationship with the regulation of gene expression in this grass is unknown. In this work sequences of small RNAs from cells chlorophyll Bouteloua gracilis were isolated and characterized, with the aim of identifying miRNAs. In addition, an in silico analysis was carried out to identify potential target mRNAs of miRNAs, as well as a search of the biological processes in which these genes involved was performed.
Material and methods
Growth of chlorophyll cells of B. gracilis
The cell line was developed in INIFAP-Celaya13. An inoculum of chlorophyll cells Bouteloua gracilis (HBK) Lag added. ex Steud., TIANSJ98, 25 ml of liquid medium MPC, the basal medium containing Murashige-Skoog plant, 2,4-D, BAP, adenine and sucrose at pH 5.8. Cultures were maintained under stirring at 90 rpm, with continuous fluorescent light (77 μmol-1 m-2) at a temperature of 33 ± 1 °C for 5 d. Subsequently, cells were harvested by filtration and the excess medium was removed. They were frozen in liquid nitrogen and stored at -70 °C until used.
Extraction of total RNA
Approximately 150 mg of B. gracilis cells were lysed by maceration with the Sample Grinding Kit™ (GE Healthcare, Sweden) system and subsequently RNA was isolated using the extraction method Trizol™ (Ambion, USA) and Plant RNA Isolation Kit™ (Ambion, USA) according to the manufacturers specifications.
Cloning of sRNAs
The cloning of the sRNAs by concatemerization was performed with the miRCat™ (Integrated DNA Technologies, USA) kit according to the manufacturer’s recommendations. Concatemers obtained were cloned into the PCR 4-TOPO (Invitrogen™, USA) vector for later insertion into chemically competent bacteria E.coli TOP10 One Shot™ (Invitrogen, USA). From the transformed cells, by extraction of plasmid DNA by the alkaline lysis miniprep was performed. SRNAs sequences inserted at the site of the polylinker of four PCR-TOPO vector was amplified with T7™ (5’-TAA-TAC-GAC-TCA-CTA-TAG-GG-3’) and T3™ (5’ oligonucleotides -ATT-AAC-CCT-CAC-TAA-GA-AGG-3’) flanking the cloning sites of the vector. The amplification program used a 94 °C initial denaturation for 1 min, 35 cycles of denaturation at 94 °C for 1 min, alignment at 55 °C for 1 min, extension at 72 °C for 1 min and at the end of the cycles, a final 72 °C extension for 10 min. The PCR products were separated on agarose gel 1.5% and the bands 190 to 400 base pair (bp) were recovered from the gel and purified using minicolumns by Wizard SV Gel and Clean- Up System according to fabricant specifications. The purified DNA was amplified and sent for synthesis and DNA sequencing at the Institute of Biotechnology of the UNAM located at Cuernavaca, Morelos, Mexico. Sequencing was performed on a Perkin Elmer Model 3730 (Applied Biosystems, USA).
Bioinformatics analysis
The obtained sequences were downloaded for FASTA alignment analysis and by using the BLASTn program through NCBI platform (http://blast.ncbi.nlm.nih.gov/Blast.cgi) with the algorithm for alignment two or more sequences, and preset parameters for an alignment of high similarity (MegaBLAST). Sequences corresponding to vector were eliminated, as well as the connector (5'-ACC-TTG-CCT-GTG-ACA G-3') that are present in sequences possible miRNAs.
The candidate sequences of 18 to 25 nucleotids were analyzed in the miRBase14 database and compared with mature miRNAs Viridiplantae kingdom there reported, according to the algorithm and preset parameters. Candidate sequences were also analyzed in the UEA Workbench15 with platform miRCat tool for the precursor sequence and secondary structure. They were used as reference genomes of Oryza sativa, Zea mays, Sorghum bicolor and Glycine max with preset parameters. Moreover, all sequences were compared to the Rfam16 database for possible annotations within the different types of RNAs (rRNAs, tRNAs, mRNAs, ncRNAs, etc.) with preset parameters.
Finally, the sequences similar to known miRNAs in order to find target mRNA was carried out using the psRNATarget17 program. Because the genome of Bouteloua gracilis has not been reported, the database annotation of transcripts rice MSU version 718 was used. The parameters used for searching for target mRNAs were more restrictive than the default values, “E value” (maximum expectation) maximum used was 2.0 (3.0 default) and “UPE” value (accessibility to the binding region) up to 20 (25 default); by decreasing the maximum values of both parameters fewer false positives are obtained17. Sequences of potential target genes were downloaded, and functional analysis of these, with Blast2GO program19 was performed.
Results
Identification of miRNAs in Bouteloua gracilis
Ten (10) plasmids were amplified, four were selected to generate PCR products ranging from 400 to 150 nucleotides. Sequencing and analysis of the amplified DNA showed the existence of nine sequences with lengths of 18 to 25 nucleotides of the miRNAs characteristic (Table 1). These sequences were analyzed on the basis of data Rfam observed lack of similarity with reported there RNAs (rRNA, tRNA snRNA, snoRNA). Analysis of sequence homology with the miRNAs deposited in miRBase14 showed that sequences 1 to 4 had the best alignment with the miRNAs miR7828-5p of Populus trichocarpa20; the miR319 reported in Brachypodium distachyon21, Oryza sativa22, Sorghum bicolor23 and Zea mays24; the miR397-3p from Malus25 and the miR397- 5p reported in Linum usitatissimum26 and Brachypodium distachyon23. However, five to nine sequences did not show similarity to previously reported miRNAs.
Table 1 In silico analysis of sRNAs sequences obtained from B. gracilis concatemers
| No | Sequence 5'-3' | Lg | miRBase | Species | ||
| 1 | GATCAGATGGAGGCTAAAATC | 21 | miR7828-5p | Ptr | ||
| 2 | AGGGAGCACCCTTCAGTCCAA | 21 | miR319 | Bdi Osa Sbi Zma | ||
| 3 | TTTCATCAACGCTGCACTCAA | 21 | miR397-3p | Mdm | ||
| 4 | ATTGAGTGCAGCGTTGATGAA | 21 | miR397-5p | Lus Bdi | ||
| 5 | ACCAACTAACCTTAGGCA | 18 | NF | NA | ||
| 6 | AAACCCCAGTTTCACACCTCCGCT | 24 | NF | NA | ||
| 7 | GTACACACATCGATCAGTCTTATT | 24 | NF | NA | ||
| 8 | TGTACATCACTAAGGTAACAAAA | 23 | NF | NA | ||
| 9 | CAATGACATCGCAAACAAGTGCCT | 24 | NF | NA | ||
MiRBase database14 was used to identify mature miRNAs.
Lg= Length of the sequence; Ptr= Populus trichocarpa; Bdi= Brachypodium distachyon; Osa= Oryza sativa; Sbi= Sorghum bicolor; Zma= Zea mays; Mdm= Malus domestica; Lus= Linum usitatissimum.
NF=Not found; NA= Not apply.
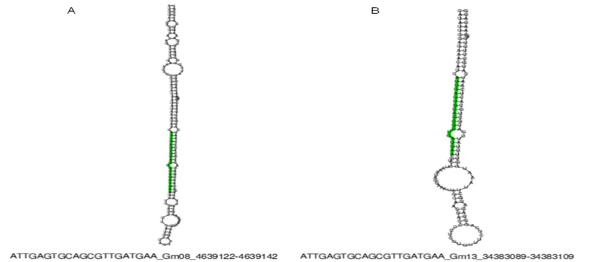
In order to characterize these sequences, the UEA miRCat Workbench tool15 was used which is a platform for predicting new miRNAs, as well as for performing mapping studies of potential miRNAs in a genome of interest. If the mapping of such genome is positive, it allows for determining whether the miRNA precursor sequence has a stem -loop (pre -miRNAs) secondary structure. The analysis was performed with all the obtained sequences and the results demonstrated that only miRNAs and characterized (miR397 and miR319) generated pre-miRNAs. So the sequence two (miR319) was located in the genomes of Zea mays, Oryza sativa and Sorghum bicolor where the structure of the pre- miRNA (Table 2, Figure 1) was obtained. With respect to sequence four (miR397-5p) it was also possible to identify their secondary structure, but only in the genome of Glycine max (Table 2; Figure 2). Regarding sequences one, five, six, seven, eight and nine, none could be identified in any of the reference genomes.
Table 2 In silico analysis of sRNAs sequences obtained from concatemers of B. gracilis, with miRCat platform15, with the genomes of soybean, corn, sorghum and rice
| Spe | Loc | Start | End | St | Sec 5'-3' | Lg | Lg Pre | % G/C | MFE |
| Zma | chr3 | 210558024 | 210558044 | - | AGGGAGCACCCTTCAGTCCAA | 21 | 170 | 54 | -66.5 |
| Osa | Os01g46984 | 541 | 561 | - | AGGGAGCACCCTTCAGTCCAA | 21 | 192 | 50 | -80.1 |
| Sbi | chro_3 | 1240353 | 1240373 | - | AGGGAGCACCCTTCAGTCCAA | 21 | 200 | 56 | -94.3 |
| Gma | Gm08 | 4639122 | 4639142 | - | ATTGAGTGCAGCGTTGATGAA | 21 | 179 | 39 | -67.3 |
| Gma | Gm13 | 34383089 | 34383109 | - | ATTGAGTGCAGCGTTGATGAA | 21 | 136 | 37 | -56.4 |
Spe= Species; Loc= location in the genome; Start= Location of start of sequence in the genome; End= Location of end of the sequence in the genome; St= string where sequence is located: Sec 5'- 3‘= sequence in the 5’ to 3 ‘ direction; Lg= length of the sequence; Lg Pre= length precursor sequence; %G/C= percentage of the content of guanine and cytokines in the pre-miRNA sequence; MFE= minimum free energy of the structure of pre-miRNA; Zma= Zea mays; Osa= Oryza sativa; Sbi= Sorghum bicolor; Gma= Glycine max; Gm08= Cromosome 8; Gm13= Cromosome 13; chro_3= Cromosome 3; Os01g46984= location number of the rice genome sequence.
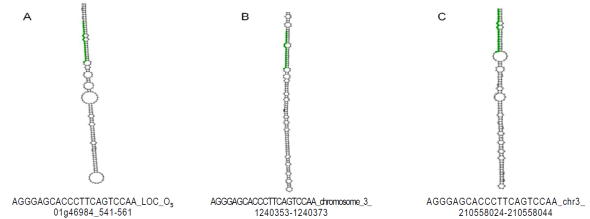
Figure 1 Secondary structure bioinformatically predicted of pre-miRNAs for sequence two (miR319-3p). Green indicates the miRNA sequence identified. A) Location of chromosome one of Oryza sativa. B) Location of chromosome three of Sorghum bicolor. C). Location of chromosome three of Zea mays
MRNA target identification
Using the psRNATarget17 program, predicted target mRNAs were used to identify potential targets of all sequences based on the degree of complementarity and the ability to access the complementary region. The analysis showed that only miR397-5p predicted six possible target mRNAs, which encode proteins laccases (Table 3). Functional analysis of target genes showed that encode proteins involved in metabolic processes and catabolic processes of lignin phenylpropanoids. In addition to these functions, each gene has different annotation involved in other processes. Five (Os01g63190, Os01g63200, Os05g38420, Os11g48060 and Os12g15680) have copper binding annotation. Two (Os01g63200 and Os12g15680) participate in the development of the reproductive system. While Os11g48060 has functions involved in cell wall formation and Os05g38420 is involved in responses due to lack of water.
Table 3 Predicted target mRNAs for four string (miR397-5p) identified with psRNATarget program17
| Name Target | Ev | UPE | Target start | Target end | Target Alignment 5'-3' | Target Description |
| Os05g38420 | 1.0 | 17.7 | 746 | 765 | UCAUCAACGCUGCACUCAAC | Protein Precursor laccase |
| Os12g15680 | 1.5 | 13.1 | 748 | 767 | UCAUCAACGCUGCGCUCAAC | Protein Precursor laccase |
| Os01g63180 | 1.5 | 17.4 | 1087 | 1106 | UCAUCAACGCUGCGCUCAAC | Precursor laccase6 |
| Os01g63200 | 1.5 | 19.7 | 665 | 684 | UCAUCAACGCUGCAGUCAAU | Protein Precursor laccase |
| Os11g48060 | 1.5 | 14.5 | 1179 | 1198 | UCAUCAACGCUGCACUGAAU | Precursor laccase 22 |
| Os01g63190 | 2.0 | 17.5 | 753 | 772 | UCAUCAACGCUGGACUCAAC | Protein Precursor laccase |
Database annotation of rice transcripts MSU version 718 was used as reference.
Ev= Expectation value; UPE= Accessibility to the target value.
Discussion
Obtaining similar sequences to mature miRNAs reported in miRBase for Bouteloua gracilis indicates that synthesizes these molecules for the regulation of their gene expression. Nine isolated sequences, only four were similar to miRNAs conserved in miRBase; while the remaining five, no records of orthologous sequences in other plant species found. Mapping analysis with miRCat program indicated miRNAs potential, with genomes of other plant species showed clear correlation of miRNAs reported in miRBase and mapping the genomes of the species in which they were identified, and the generation of stable stem -loop secondary structures; however, the rest did not show any selected genome mapping. Together, these results confirm the high degree of conservation of miRNAs, miR319 and miR397 found in grasses such as Zea mays, Oryza sativa and Sorghum bicolor, in addition to the legume Glycine max, as well as in B. gracilis. It is interesting that although the number of sequences obtained is low, it was possible to detect the presence of both miR397-5p since it is complementary to miR397 -3p, so this miRNA might be active in B. gracilis. Regarding sequences five to nine, the absence of orthologs and the negative to one of the reference genomes employees mapping indicates the possibility that these sequences are specific to B. gracilis. Regarding sequence one, although it was similar with miR7828-5p of Populus trichocarpa, it was very low, since it had five mismatches.
The detected potential target mRNAs were predicted from the sequence four (miR397-5p); therefore, it is possible that the expression of this miRNA negatively regulates expression of laccases, which are glycoprotein enzymes having the function of oxidizing phenolic compounds present in the lignin27 . Inhibition of expression of these enzymes may be due to the fact that, as the chlorophyll cells are undifferentiated, in which there is the presence of lignin, the presence of laccases oxidize to such phenolic compounds are required. On the other hand, in the absence of lignin present, laccases could oxidize other phenolic compounds, which could affect the proper functioning of cells. Further, these genes had also functional annotations involved in the development of the reproductive system, formation of cell wall and response to lack of water, so it is expected that these genes are being muted because it is possible that none of these biological processes occur in undifferentiated cells, from which this study was done. On the other hand, miR319 is a much characterized miRNA, and although it was not possible predict a target in this analysis, it has been reported that one of its main objectives is in TCP transcription factors class II; which participate in the negative regulation of cell growth as reported in Arabidopsis28.
The fact that other sequences did not present potential targets does not mean they do not exist; the above, it may be due to the fact that the selected parameters for analysis were very high to avoid false positives, or that targets are specific sequences of B. gracilis.
Conclusions and implications
In this study it was possible to identify four miRNAs reported in miRBase; two of them are highly conserved. These results confirm the presence of small RNAs and miRNAs in B. gracilis. The identification of miR397- 5p, miR397 -3p, as well as their potential target mRNAs that encode for enzymes with laccase activity, is an indication that this molecule is very active in these cells. The evaluation of this miRNA expression in different plant tissues and cells and plants grown under conditions of water stress, will reveal the physiological relevance of this miRNA in B. gracilis.
Acknowledgments
Luz Angelica Muñoz for technical help.
REFERENCES
1. Mallory AC, Vaucheret H. Functions of microRNAs and related small RNAs in plants. Nat Genet 2006;38:S31-S36. [ Links ]
2. Jung JH, Seo PJ, Park CM. MicroRNA biogenesis and function in higher plants. Plant Biotechnol Rep 2009;3:111-126. [ Links ]
3. Luan F, Han Y, Zhu H, Shao Y, Chen A, Tian H, Luo Y, Zhu B. Computational predicting novel microRNAs in tomato and validating with RT-PCR. Russian J Plant Physiol 2010;57:469-479. [ Links ]
4. Guzman F, Almerao MP, Körbes AP, Loss-Morais G, Margis R. Identification of microRNAs from Eugenia uniflora by high-throughput sequencing and bioinformatics analysis. PLoS ONE 2012;7:e49811. [ Links ]
5. Jones-Rhoades MW. Conservation and divergence in plant microRNAs. Plant Mol Biol 2012;80:3- 16. [ Links ]
6. Zeng C, Wang W, Zheng Y, Chen X, Bo W, Song S, Zhang W, Peng M. Conservation and divergence of microRNAs and their functions in Euphorbiaceaous plant. Nucleic Acid Res 2009;38:981-995. [ Links ]
7. Ha M, Pang M, Agarwal V, Chen ZJ. Interspecies regulation of microRNAs and their targets. Biochim Biophys Acta 2008;1779:735-742. [ Links ]
8. Evertson J. Bouteloua gracilis. American Nurseryman. 2010. http://www.amerinursery.com. Acesessed Jun 15, 2014. [ Links ]
9. Valdés-Reyna J, Dávila APD. Clasificación de los géneros de gramíneas (Poaceae) mexicanas. Acta Bot Mexicana 1995;33:37 -50. [ Links ]
10. Aguiar M, Lauenroth W. Local and regional differences in abundance of co-dominant grasses in the shortgrass steppe: a modeling analysis of potential causes. Plant Ecol 2001;156:161-171. [ Links ]
11. Ortiz MV. Fluctuación en el contenido de proteína cruda y fósforo en 3 ecotipos de zacate navajita (Bouteloua gracilis) y banderilla (Bouteloua curtipendula) y 2 ecotipos de zacate toboso (Hilaria mutica) en cuatro estados fenológicos. Contenido y fluctuación de nutrientes de las especies forrajeras consumidas por el ganado en los agostaderos de Chihuahua. Boletín Pastizales RELC-INIP-SARH. 1984;15:1-20. [ Links ]
12. Morales-Nieto C, Madrid-Pérez L, Melgoza-Castillo A, Martínez-Salvador M, Arévalo-Gallegos S, Rascón-Cruz Q, Jurado-Guerra P. Análisis morfológico de la diversidad del pasto navajita (Bouteloua gracilis Willd ex Kunth) Lag ex Steud. en Chihuahua México. Téc Pecu Méx 2009;47:245-256. [ Links ]
13. Aguado-Santacruz GA, Cabrera-Ponce JL, Ramírez-Chávez E, C. León-Ramírez G, Rascón-Cruz Q, Herrera-Estrella L, Olalde-Portugal V. Establishment, characterization and plant regeneration from highly chlorophyllous embryogenic cell cultures of blue grama grass, Bouteloua gracilis (H.B.K) Lag. Ex Steud. Plant Cell Rep 2001;20:131-136. [ Links ]
14. Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep sequencing data. Nucleic Acids Res 2011;39:D152-D157. [ Links ]
15. Stocks MB, Moxon S, Mapleson D, Woolfenden HC, Mohorianu I, Folkes L, Schwach F, Dalmay T, Moulton V. The UEA sRNA workbench: a suite of tools for analysing and visualizing next generation sequencing microRNA and small RNA datasets. Bioinformatics 2012;28:2059-2061. [ Links ]
16. Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 2012;41. doi:10.193/nar/gks1005. [ Links ]
17. Dai X, Zhao PX. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res 2011;W155 -W159. [ Links ]
18. Kawahara Y, De-la-Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, et al. Improvement of Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013;6:4-13. [ Links ]
19. Conesa A, Götz S, García-Gómez JM, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005;21:3674-3676. [ Links ]
20. Shuai P, Liang D, Zhang Z, Yin W, Xia X. Identification of drought-responsive and novel Populus trichocarpa microRNAs by high-throughput sequencing and their targets using degradome analysis. BMC Genomics 2013;14:233. [ Links ]
21. Bertolini E, Verelst W, Horner DS, Gianfranceschi L, Piccolo V, Inzé D, Pé ME, Mica E. Addressing the role of microRNAs in reprogramming leaf growth during drought stress in Brachypodium distachyon. Mol Plant 2013;6:423-443. [ Links ]
22. Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Molecular Cell 2004;14:787-799. [ Links ]
23. Dezulian T, Palatnik JF, Huson D, Weigel D. Conservation and divergence of microRNA families in plants. Genome Biol 2005;6:P13. doi:10.1186/gb -2005-6 -11-p13. [ Links ]
24. Zhang L, Chia JM, Kumari S, Stein JC, Liu Z, Narechania A, Maher CA, et al. A genome-wide characterization of microRNA genes in maize. PLoS Genet 2009:5(11). doi:10.1371/journal.pgen.1000716. [ Links ]
25. Xia R, Zhu H, An YQ, Beers EP, Liu Z. Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biology 2012;13:R47. doi:10.1186/gb-2012-13-6-r47. [ Links ]
26. Barvkar VT, Pardeshi VC, Kale SM, Qiu S, Rollins M, Datla R, et al. Genome-wide identification and characterization of microRNA genes and their targets in flax (Linum usitatissimum). Planta 2013;237:1149-1161. [ Links ]
27. Preussler CA, Shimizu E, Villalba LL, Zapata PD. Inducción con cobre de la enzima lacasa en el hongo de pudrición blanca Trametes villosa (sw.:Fr.) Kreisel. Rev Cienc Tecnol 2009;12:9-16. [ Links ]
28. Nag A, King S, Jack T. miR319a targeting of TCP4 is critical for petal growth and development in Arabidposis. PNAS 2009;106(52)22534-22539. [ Links ]
Received: May 20, 2015; Accepted: September 19, 2015
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
