










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista mexicana de ciencias pecuarias
versión On-line ISSN 2448-6698versión impresa ISSN 2007-1124
Rev. mex. de cienc. pecuarias vol.3 no.2 Mérida abr./jun. 2012
Revisión de literatura
Toxemia de la gestación en ovejas. Revisión
Ewe pregnancy toxemia. Review
Luis Cal–Pereyraa, Jorge Acosta–Dibarratb, Alejandro Benechc, Stella Da Silvac, Andrea Martína, José Ramiro González–Montañad
a Departamento de Patología, Facultad de Veterinaria, Universidad de la República, Montevideo, Uruguay.
b Centro de Investigación y Estudios Avanzados en Salud Animal, Facultad de Medicina Veterinaria y Zootecnia, Universidad Autónoma del Estado de México, Km. 15.5 Carretera Panamericana Toluca–Atlacomulco, Toluca, Estado de México, 50200, Toluca, México. Teléfono y Fax: 01(722)296 55 55 y 296 89 80. jpacosta00@hotmail.com. Correspondencia al segundo autor.
c Departamento de Fisiología, Facultad de Veterinaria. Universidad de la República. Montevideo, Uruguay.
d Departamento de Medicina y Anatomía, Facultad de Veterinaria. Universidad de León, León, España.
Recibido el 26 de mayo de 2010.
Aceptado el 18 de agosto de 2010.
Resumen
En este trabajo se presenta una exhaustiva revisión de la toxemia de la gestación en la oveja. La misma ha sido dividida en diferentes secciones para lograr una mejor comprensión de los factores que predisponen a esta enfermedad, su patogenia, signos clínicos, los hallazgos postmortem y el diagnóstico. Por último se presenta un análisis de los distintos tratamientos que se utilizan en esta enfermedad.
Palabras clave: Ovinos, Toxemia de la gestación, Cuerpos cetónicos, Glicemia, Esteatosis hepática, Patogenia.
Abstract
In this work a comprehensive review of ewe pregnancy toxemia is presented. Different thematic sections were considered, for a better understanding of the predisposing factors involved, their pathophysiology, clinical signs, post mortem findings, and the diagnosis. Finally, an analysis of current therapy of the disease is presented.
Key words: Sheep, Pregnancy toxemia, Ketone bodies, Glycaemia, Hepatic steatosis, Phathogenesis.
INTRODUCCIÓN
La toxemia de la gestación es un trastorno metabólico que afecta a las ovejas preñadas durante el último tercio de la gestación, especialmente en las últimas seis semanas, como consecuencia de la incapacidad del organismo para mantener la homeostasis energética al enfrentarse en esta etapa, a un balance energético negativo(1,2).
La referencia más antigua de esta enfermedad probablemente es la de Seaman, quien, en 1854 reseñó una serie de muertes de ovejas atribuibles a la toxemia de la gestación(3). Esta patología ha recibido otras denominaciones entre las que se destacan: fiebre de la oveja parturienta, parálisis del preparto, estercoremia, acidosis de la oveja gestante, cetosis ovina, acetonemia, paresia o eclampsia anteparto, hepatitis parenquimatosa aguda, enfermedad del hígado blanco, enfermedad de la preñez, enfermedad de los gemelos, enfermedad de la oveja gestante, toxemia gravídica y toxemia de la preñez(3,4,5). Esta enfermedad metabólica se presenta también en otras hembras en gestación avanzada como la cabra y la cierva(1,6,7) y muy raramente en la vaca(8).
Generalmente se ha realizado el estudio de esta patología en animales que presentaron espontáneamente la enfermedad(6,8), pero gracias a su inducción experimental mediante ayuno, restricción alimenticia o inyección de D–BOHB (D–ß hidroxibutirato) se han logrado alcanzar enormes progresos en el conocimiento de la misma, y principalmente de su patogenia(1,9).
FACTORES PREDISPONENTES
Las causas que predisponen a esta patología se pueden clasificar en tres grandes grupos; nutricionales, estresantes e inherentes al animal (3,4):
Nutricionales: La subnutrición y el ayuno al final del período de gestación conducen a las ovejas a un balance energético negativo y las predispone a la enfermedad. La subnutrición produce una intensa hipoglucemia, que ocasiona la disminución del flujo de la glucosa uterina, umbilical y uteroplacental (10). Por otro lado, la sobrealimentación en los primeros meses de gestación, puede ocasionar la posterior disminución voluntaria de la ingesta al reducirse la capacidad ruminal por el depósito de grasa intra–abdominal y por el aumento del volumen del útero gestante en las últimas semanas de la gestación(6).
Andrews(11) sugiere que esta patología se ve generalmente en rebaños de parición temprana, lo que ocurre en pleno invierno y conduce a un fallo gradual en el plano nutricional, seguido de un período de ayuno o estrés, bien sea como consecuencia del manejo o del clima.
Estresantes. Los factores estresantes son aquéllos que originan un mayor gasto de energía, promoviendo el consumo de las reservas energéticas y una disminución de la ingesta de alimento. Entre estos factores se destacan las condiciones climáticas adversas (frío, lluvias intensas, heladas, granizo) asociadas a una inadecuada protección y abrigo, o cuando estas mismas condiciones impiden a las ovejas salir a pastar, obligando un cambio en su alimentación(4,6,11).
Inherentes al animal. Dentro de éstas, la gestación múltiple es una de las principales, debido a las altas demandas energéticas de los fetos y al importante aumento del volumen del útero. La oveja en estas condiciones reduce sus movimientos, disminuyendo así el consumo de alimentos. En países como Uruguay, donde la oveja se cría en condiciones de campo natural, se puede presentar en ovejas con gestaciones simples, principalmente en inviernos rigurosos con grandes carencias nutricionales(4).
Causas como la edad avanzada de las madres, la mala dentición y los procesos podales también inciden negativamente, limitando el consumo de alimentos. Las parasitosis gastrointestinales y hepáticas (fasciolasis hepática y quiste hidático) o cualquier otra hepatopatía que provoque insuficiencia hepática, deben asimismo ser consideradas(3,4).
Wastney et al(12) proponen que existe una gran variabilidad individual en la susceptibilidad a enfermar después de someterlas a ayuno en el período final de la gestación. Estos autores encontraron una alta resistencia a la insulina en los tejidos periféricos de las ovejas susceptibles. En este sentido, recientemente se determinó que la malnutrición de las ovejas al final de la gestación condiciona la respuesta de las corderas a los desafíos metabólicos en su vida adulta, como lo son la gestación y la lactación (13).
Para Duffield(14) y Rook(6) la susceptibilidad individual podría estar relacionada con diferencias genéticas y para Duehelmeier et al(15) la raza es un importante factor determinante en la resistencia a desarrollar la enfermedad.
PATOGENIA
En la Figura 1 se esquematizan los procesos metabólicos que llevan a la Toxemia de la gestación.
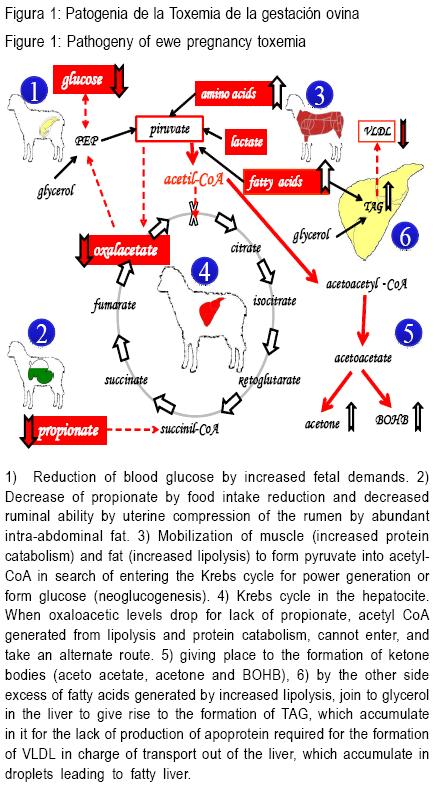
La causa determinante de esta patología es una alteración del metabolismo energético, fundamentalmente en los mecanismos que participan en la homeostasis de la glucosa(5). Aunque la etiopatogenia no es completamente conocida, esta enfermedad es esencialmente una forma severa de cetosis, caracterizada por una baja circulación de glucosa en sangre y altos niveles de cuerpos cetónicos(6,11).
Sigurdsson(5) propone que el balance entre alimentación y requerimientos es el elemento central en la patogenia de la toxemia de la gestación, a lo que Sargison et al(8) agregan que la enfermedad es el resultado de un fallo en la energía proporcionada por la dieta y la proveniente de la neoglucogénesis (NG) para cubrir una demanda fetal incrementada de glucosa, conduciendo a las ovejas a un balance energético negativo.
Los requerimientos energéticos en la gestación avanzad a aumentan sobre los niveles de mantenimiento en aproximadamente 150 % en ovejas con una gestación simple y hasta 200 % en ovejas gestando mellizos. Este incremento es causado por el hecho de que cerca del 85 % del crecimiento fetal ocurre durante esta etapa, aumentando el drenaje fetal de glucosa(6). El requerimiento feto–placentario de energía puede llegar a representar el 45 % de la glucosa materna y el 72 % de la oferta de aminoácidos maternos(7,16).
El ingreso de glucosa al feto parece ser independiente de la regulación de la glucosa materna, ya que es relativamente insensible a la concentración sérica de insulina(7). A pesar que los niveles de glucosa sanguínea en la oveja declinen y se ocasione un perjuicio en la madre, los mecanismos de protección en el suministro de glucosa al feto aseguran la viabilidad de éste a corto plazo, que ocurre a expensas de la homeostasis de la glucosa en la madre(6).
A este aumento espectacular de los requerimientos de energía se agrega una disminución de la ingesta de materia seca, fundamentalmente a causa de una capacidad ruminal reducida como consecuencia del aumento de volumen del útero en gestación(6,11).
Cuando el ingreso de alimento se vuelve restrictivo, disminuye la concentración de glucosa en sangre. La severidad de esta hipoglucemia depende del número de fetos, siendo mayor en aquellas ovejas que gestan más de un feto(17). La teoría más aceptada para explicar la patogenia de esta enfermedad, propone que fracasan los mecanismos de regulación de la homeostasis energética en animales con un balance energético negativo, lo que resulta en una incapacidad para mantener la concentración de la glucosa en sangre, y la producción desenfrenada de cuerpos cetónicos(6,8). Esto último se produce por una excesiva síntesis de cuerpos cetónicos y por una disminución en su utilización(8). De este modo parece que en algún punto, los mecanismos que contribuyen a mantener la homeostasis de los carbohidratos y el suministro de energía son remplazados por un sistema destructivo de retroalimentación negativo, que da lugar a hipoglucemia y a un rápido incremento de la concentración de cuerpos cetónicos en sangre(18).
Uno de los factores determinantes más importantes de que una oveja sufra o no la enfermedad, sería la funcionalidad hepática, ya que el hígado es el regulador de la concentración de la glucosa sanguínea y del aporte de glucosa a los tejidos, y prácticamente es el único órgano donde se realiza la NG, a pesar de que existen pequeños aportes del riñón(3,7).
Se ha demostrado que el ayuno a partir del día 130 de gestación durante un máximo de seis días permite reproducir un cuadro clínico de toxemia de la gestación en ovejas Corriedale que portan un solo feto(19), demostrándose asimismo que el ayuno provoca una rápida movilización de TAG desde el tejido adiposo, lo que se ve reflejado en el rápido ascenso de los valores de los NEFA, representando el cambio sanguíneo más precoz en las ovejas sometidas a ayuno. Este incremento de la movilización lipídica provoca una alta incidencia de esteatosis hepática en estos animales(19). La correlación positiva encontrada entre la actividad sérica de la enzima aspartato aminotransferasa (ASAT) y el grado de vacuolización hepática indica que esta enzima podría ser un indicador precoz y fiable del daño hepático en ovejas con Toxemia de la gestación clínica(19).
El aumento desproporcionado de cuerpos cetónicos en sangre conduce a una acidosis metabólica, ya que estos se comportan como ácidos fuertes (pK=4). Asimismo, el paso de estos cuerpos cetónicos ionizados a la orina promueven la eliminación de ciertos cationes (Na+, K, Ca++), determinando una expoliación electrolítica y deshidratación, que puede conducir a un fallo renal. El acetoacetato tiene asimismo un efecto directo sobre el cerebro, provocando una disminución en el consumo cerebral de O2 y síntomas neurológicos. Otra causa de los signos nerviosos presentes en esta enfermedad probablemente sea una encefalopatía hipoglucémica(4,7).
SIGNOS CLÍNICOS
Gran parte de los signos clínicos se explican por la intensa hipoglucemia que sufre la oveja con toxemia de la gestación. Cuando los niveles de glucosa en sangre descienden desde 50 a 70 mg/dl (valores considerados fisiológicos para esta especie), hasta 20 mg/dl, se produce una encefalopatía hipoglucémica con lesiones cerebrales irreversibles, siendo la causante de la sintomatología nerviosa de esta enfermedad(5,19,20). La depresión del metabolismo neuronal se intensifica por el efecto directo que posee el acetoacetato de disminuir el consumo cerebral de oxígeno(3,4). El comienzo de las manifestaciones clínicas es relativamente brusco, aunque es probable que la enfermedad se venga desarrollando desde tiempo atrás, en forma subclínica(4).
En las etapas iniciales de la enfermedad los leves signos clínicos a menudo pasan inadvertidos, los animales afectados se muestran apáticos y lentos, alimentándose cerca del rebaño(6). A medida que la patología progresa, las ovejas afectadas se retrasan y se separan del resto del rebaño, perdiéndose frecuentemente. Al aumentar el grado de depresión no reaccionan ante la presencia del hombre o de perros, sufren la pérdida de reflejos auditivos y oculares, la marcha se torna dificultosa chocando contra objetos y avanza en círculos. Algunos animales tienen tendencia a permanecer inmóviles presionando la cabeza contra los objetos. Durante esta fase inicial el animal disminuye la ingesta de alimentos y agua, mostrando constipación con excremento duro y seco(4,6,19). A medida que la enfermedad progresa la debilidad y la depresión aumentan, la oveja adopta posturas anormales de la cabeza como la de "contemplar las estrellas" (7) o la posición menos frecuente de "perro sentado"(3), siendo común el rechinar de los dientes debido a movimientos reflejos de la mandíbula y la lengua(4,19).
En las últimas etapas la acidosis metabólica desarrollada incrementa la frecuencia respiratoria, se presentan frecuentemente contracciones mioclónicas de la cabeza, espalda y extremidades(3,4,19), sutiles episodios convulsivos(6,7), terminando el animal en decúbito esternal con la cabeza girada hacia el flanco; posteriormente progresa al decúbito lateral donde puede permanecer por 3 ó 4 días en un estado de depresión profunda, muriendo el 80 a 90 % de los casos no tratados(3,6).
Si se produce la muerte del o los fetos, la oveja puede presentar una recuperación transitoria al incrementarse la concentración plasmática de glucosa y, por tanto, la concentración de glucosa del fluido cerebroespinal. Sin embargo, debido a que en esta especie es difícil que ocurra la expulsión de los fetos muertos, la septicemia consecuente llevará a una depresión más profunda con aumento de la temperatura(7,14).
En caso de que la oveja que padece Toxemia de la gestación sobreviva hasta el final de la gestación, generalmente ocurre distocia, que se asocia a una pobre actividad de la musculatura uterina y abdominal y a una pobre dilatación cervical. En muchos de estos casos se presenta además retención de placenta, lo que conduce a metritis y posteriormente a la muerte(6,7). Las ovejas afectadas que se recuperan, a menudo paren un cordero muerto o pequeño y débil, el cual muere a los pocos días de nacer. Estas madres generalmente producen poca leche, sus corderos son susceptibles a la hipotermia y diarreas, y la mortalidad posparto suele ser alta(2,11). En estas ovejas el reinicio de la actividad cíclica del ovario, la respuesta ovárica y la fertilidad están muy disminuidas(21).
La toxemia de la gestación es a menudo un problema de rebaño. En los grupos afectados la enfermedad puede alcanzar proporciones de brote cuando se afectan muchos individuos todos los días durante un período de varias semanas(7).
HALLAZGOS POSTMORTEM
El estado de la canal varía de acuerdo con la condición corporal de la oveja al inicio de la enfermedad, la cual puede encontrarse en buenas condiciones o muy emaciada, presentando grandes cantidades de grasa abdominal y subcutánea(6,11). La grasa corporal puede presentar consistencia disminuida y aspecto gelatinoso, debido a la licuefacción en aquellos casos de evolución crónica, en tanto que en los de evolución aguda las áreas de licuefacción suelen estar limitadas a pequeños puntos(4).
Es muy orientativo que en animales muertos por Toxemia de la gestación, se encuentra el útero con fetos muertos y con cierto grado de autolisis, estando en estos casos rodeado de un material espeso, viscoso y negro–amarronado(4).
El hígado está generalmente agrandado, pálido, friable y con bordes redondeados; varía en color desde el rosa al amarillo anaranjado y su consistencia es grasienta al tacto. La histopatología revela infiltración grasa caracterizada por esteatosis hepatocítica microvesicular(4,6,9). Las glándulas adrenales están usualmente aumentadas de tamaño, presentando hemorragia cortical; esta hipertrofia de la glándula se corresponde con una respuesta al prolongado estímulo de la hormona adenocorticotrofina (ACTH)(6,11).
Las lesiones renales son a menudo pobremente definidas, describiéndose una degeneración glomerular e infiltración grasa del epitelio tubular renal(11). La histopatología del cerebro revela cambios como hinchamiento del núcleo de los astrocitos e hipertrofia, proliferación y necrosis neuronal cerebrocortical . Se han reportado, asimismo, necrosis de las células de Purkinje y vacuolización de la sustancia blanca cerebral y cerebelar subcortical. Estos hallazgos soportan la teoría que muchos de los signos clínicos de la toxemia de la gestación son consecuencia de una encefalopatía hipoglucemica(22).
DIAGNÓSTICO
El diagnóstico de la enfermedad es sencillo y en general no ofrece mayores dificultades siempre que se disponga y tenga en cuenta la información de la anamnesis, del examen clínico, de los exámenes colaterales y los resultados de las lesiones postmortem.
La hipoglucemia sirve de ayuda diagnóstica en las fases iniciales de la enfermedad, siendo común encontrar valores de 20 a 40 mg/dl (1.1 a 2.2 mmol/l). En casos graves de la enfermedad los niveles de glucosa sanguínea pueden llegar a descender a menos de 20 mg/dl (1.1 mmol/l)(4,19,23).
Según algunos autores, en las etapas finales puede existir normo e incluso hiperglucemia, por lo que su determinación en estas etapas posee un valor limitado(8,17,19). Este aumento de la glicemia en etapas avanzadas de la enfermedad podría estar relacionado con un aumento del cortisol sérico(19,24, 25).
La hipercetonemia es una constante en ovejas toxémicas. La magnitud de la cetonemia inducida por el ayuno o la subnutrición, depende del plano de nutrición al que fueron sometidas previamente, influencia que no se refleja en la glicemia. La mayoría de los autores proponen que es posible realizar el diagnóstico clínico de toxemia de la gestación cuando la concentración de 13–hidroxibutirato (BOHB) sérico está por encima de los 3.0 mmol/l (54.05 mg/dl), aunque comúnmente supera los 5.0 mmol/l (90,09 mg/ dl)(7,11). Pethick y Lindsay (26) sugieren que el valor diagnóstico de laboratorio del BOHB es de 2 mmol/l (36.03 mg/dl). Los niveles postmortem de BOHB en el humor acuoso por encima de 2.5 mmol/l (45.0 mg/dl) y mayores de 0.5 mmol/l (9.0 mg/dl) en el fluido cerebroespinal de ovejas, también son considerados de valor diagnóstico(27).
Se sugiere que la concentración de fructosamina sérica puede ser un indicador temprano de la enfermedad en un rebaño cuando la concentración de BOHB está en límites normales. En este sentido se describe que los animales afectados presentan una concentración sérica de fructosamina marcadamente más baja que la de las ovejas normales, lo que sugiere una persistente hipoglucemia en estos animales(28).
La valoración de la acidosis metabólica puede realizarse mediante la determinación del pH urinario, el cual puede ser ácido, llegando incluso a un pH 5(29). No obstante, se ha apreciado que no existe diferencia significativa en los valores del pH urinario en las ovejas con y sin síntomas, por lo que se podría deducir que el pH urinario no guarda relación con la presentación de síntomas clínicos(19).
Los niveles de cortisol sérico están elevados(19,25), sugiriéndose que concentraciones de cortisol sérico superiores a 10 ng/ml son indicativas de toxemia de la gestación clínica(25).
Se menciona que en las últimas etapas de la enfermedad puede aparecer una disfunción renal con aumento de la urea y creatinina sérica(3,11). La alta movilización lipídica en los animales afectados provoca una alta incidencia de esteatosis hepática, afectando la totalidad del acino hepático, siendo este acúmulo de tipo microvesicular, lo que se relaciona con el aumento de la aspartato aminotransfereasa (ASAT)(9).
Como consecuencia de la disfunción hepática y renal se presenta disminución de la albúmina sérica, aumento de las enzimas indicadoras de daño hepático como ASAT, alanino aminotransferasa (ALT) y gamma glutamiltransferasa (GGT)(30). La correlación positiva encontrada entre la actividad sérica de la ASAT y el grado de vacuolización hepática, indican que esta enzima podría ser usada como un indicador precoz y fiable del daño hepático en ovejas con toxemia de la gestación clínica(9).
Las ovejas con toxemia de la gestación presentan incremento en los niveles de interleucina 1 beta (IL–1b), factor de necrosis tumoral (TNFa) y proteína quimiotáctica de monocitos –1 (MCP–1) que podrían servir como potenciales indicadores del pronóstico de la enfermedad, sin embargo faltaría determinar su importancia en la patogenia de la misma(31).
TRATAMIENTO
Los tratamientos descritos para la toxemia de la gestación tienen resultados variables y resultan ser costosos cuando la enfermedad afecta a un número elevados de animales.
La respuesta al tratamiento es efectiva, si éste se instaura en los primeros estadios de la enfermedad, cuando no se han establecido lesiones neurológicas irreversibles y cuando el animal aún no está en decúbito(3,4,7). Por ello es muy importante el diagnóstico precoz de la enfermedad, ya que permitirá una terapéutica racional y efectiva(19).
El objetivo prioritario en el tratamiento de la enfermedad es el aumento de la formación de glucosa y su utilización a nivel tisular, debiendo incrementar también la utilización de los cuerpos cetónicos(3), el combate a la acidosis y los trastornos hidroelectrolíticos(4,7). La normalización de la glucemia y los cuerpos cetónicos en sangre llevará a restablecer el apetito normal(18). Esto sería posible lograrlo abordando uno o más objetivos a la vez. El metabolismo lipídico puede ser alterado por la supresión de la movilización de los ácidos grasos desde el tejido adiposo, reduciendo el ingreso de estos al hígado, y con ello, su transporte al interior de las mitocondrias hepáticas. La concentración de glucosa sanguínea se puede aumentar reduciendo su utilización o estimulando la NG.
Glucosa. El tratamiento de esta patología con soluciones de glucosa o dextrosa es una de las terapias más comunes. Puede ser efectivo en etapas tempranas de la enfermedad, antes de la aparición del daño cerebral irreversible o de severas complicaciones secundarias. Se han propuesto varias alternativas : a) suero glucosado al 5 % en dosis de 250 a 500 ml aplicados por vía intraperitoneal o intravenosa en las primeras etapas de la enfermedad(4), b) suero glucosado isotónico al 5 a 10 % a dosis de 250 a 1000 ml aplicados por vía intravenosa o intraperitoneal, al menos dos veces al día debido a la rápida metabolización de la glucosa(3) y c) soluciones hipertónicas de glucosa, que deben utilizarse con precaución, ya que pueden aumentar el riesgo de acidosis metabólica(3,4). En casos avanzados de la enfermedad aparece hiperglucemia y en este momento la terapia a base de glucosa no sería recomendable(19,32).
La infusión de glucosa conduce a una reducción de la NG, caída de la concentración sérica de los cuerpos cetónicos, y disminución de los NEFA plasmáticos. Esta respuesta es probablemente debida a la reducción de la lipólisis. El principal efecto de la infusión de glucosa en el metabolismo hepático parece ser el de disminuir la cetogénesis(18).
Se debe tener presente que la inyección intravenosa rápida de glucosa resulta en una hiperglucemia pasajera, conduciendo a un aumento de la diuresis y pérdida urinaria de la mayor parte de la glucosa administrada(18).
Algunos autores(4,33) afirman que el tratamiento con glucosa intravenosa debería ir siempre unida a la administración parenteral de insulina, debido a que ésta promueve la rápida utilización de la glucosa, ayudando a superar el efecto antagonista que el cortisol tiene sobre la insulina endógena y el metabolismo de la glucosa.
El beneficio de la infusión de glucosa como terapia para el hígado graso no está suficientemente establecido. El tratamiento con insulina reduce la magnitud de la lipólisis del tejido adiposo, lo cual es beneficioso. Sin embargo reducir la magnitud del ingreso de ácidos grasos a la mitocondria hepática es más importante, pues éste sería directamente el camino de los ácidos grasos para la esterificación y acumulación en las células hepáticas. Suplementar la terapia con insulina sería más racional en el tratamiento del hígado graso, porque la insulina estimula la secreción hepática de lipoproteínas(18).
Azucares distintos de la glucosa. Otros azucares distintos de la glucosa han sido usados en la terapia de la cetosis, como por ejemplo la fructosa y el sorbitol. Estas sustancias son metabolizadas a glucosa, y la ventaja principal sobre ella es que son utilizadas por el hígado antes que por los tejidos periféricos; de esta manera la glucosa resultante del metabolismo de estos compuestos se concentra en el hígado, y suprime la cetosis de forma más efectiva que una dosis equivalente de glucosa administrada directamente, la cual es metabolizada en los tejidos extra hepáticos(18). Sin embargo hay poca información disponible sobre el uso de estos compuestos en el tratamiento de la toxemia de la gestación.
Precursores de la glucosa. Los precursores de la glucosa por vía oral, solos o combinados con alguna terapia parenteral, incluyen alglicerol, al propilenglicol (1,2 propanediol) (PG) y a las sales del ácido propiónico. En la práctica el más popular es el PG, constituyendo uno de los tratamientos de mayor difusión para la toxemia de la gestación(4,29).
El glicerol es degradado lentamente en el rumen, produciendo una elevada proporción de propionato, principal precursor de la glucosa, dando como resultado una elevación de la glucemia por un período relativamente prolongado (4,34), con una brusca reducción de cuerpos cetónicos en plasma y en orina(29).
El PG es administrado en forma oral y se absorbe directamente desde el rumen, aproximadamente a una tasa de un 40 % por hora, con una vida media de 3 h(35). En animales a los cuales se les administra por primera vez el PG, la mayoría se absorbe intacto, y sólo una pequeña cantidad es metabolizada a ácido propiónico; sin embargo tras una prolongada administración, la magnitud del metabolismo ruminal es probable que se incremente(18).
Los niveles máximos de PG en sangre ocurren dentro de los 30 min posteriores a su administración, y la máxima conversión a glucosa sanguínea ocurre alrededor de 4 h después de su aplicación, probablemente mediante su conversión a piruvato, con la subsiguiente producción de oxalacetato vía piruvato carboxilasa. El incremento del oxalacetato disponible es de esperar que conduzca a un incremento de la síntesis de citrato, suprimiendo de esta forma la cetogénesi s(18).
La eficacia de estos compuestos son de utilidad siempre que no exista un severo compromiso del hígado(4,6), ya que la capacidad de utilizar PG estaría reducida cuando existe infiltración grasa. En animales sospechosos de tener una severa esteatosis hepática, el PG debe ser usado con cuidado, ya que en grandes dosis puede acumularse y contribuir fuertemente a la aparición de depresión y somnolencia(18).
La administración de un tratamiento oral a base de glicerol–propilenglicol a una dosis de 100 ml, dos veces por día, fue efectiva en la normalización de los parám etros metabólicos tras la inducción experimental de la enfermedad mediante ayuno. Sin embargo, en condiciones prácticas el tratamiento puede presentar fallos como consecuencia de la severidad y duración del caso, y de la presentación de diversas complicaciones como son acidosis, daño cerebral irreversible, insuficiencia renal y deshidratación(29). Es por esta razón que Roock(6) propone que el tratamiento de las ovejas más afectadas incluya la administración de 100 a 200 ml de propilenglicol vía oral, dos a cuatro veces por día, además de la inyección intravenosa de una solución de dextrosa. Esta última se puede administrar de la siguiente manera: 250 ml al 20 %, 500 ml al 10 % ó 120 ml al 50 %. Si además del glicerol o PG se administra 40 UI de insulina por vía intramuscular, la mortalidad desciende a un 50 % aproximadamente(32).
Otros autores(36) sugieren la posibilidad de administrar una solución de glucosa vía oral, después de inducir el cierre de la gotera esofágica con una dosis de lisina–vasopresina a una dosis de 0,08 UI/kg por vía intravenosa. Esto permitiría el paso de la glucosa directamente hacia el abomaso, produciendo un rápido incremento de la glicemia.
Insulina. La insulina posee un efecto antilipolítico y anticetogénico de gran interés terapéutico(37). La administración de la hormona que induce hipoglucemia para el tratamiento de una enfermedad que cursa con hipoglucemia parece paradójica, especialmente cuando es conocido que la aplicación de insulina en animales normales puede inducir signos de cetosis. Es conveniente considerar, sin embargo, que con la administración de insulina se producen solamente hipoglucemia y signos de cetosis, pero no se produce hipercetonemia. De hecho la insulina es una potente hormona anticetogénica, sugiriendo que cuando el efecto de la hipoglucemia puede ser evitado, es un buen agente terapéutico para el tratamiento de la cetosis(18). En la terapia contra la cetosis la insulina usualmente es administrada en combinación con glucosa(6,33) o con glucocorticoides, para contrarrestar su efecto hipoglucemiante(18). Marteniuk y Herdt(7) recomiendan administrar la insulina por vía subcutánea o intramuscular bajo la forma de liberación lenta a dosis de 20 a 40 UI, una vez al día durante tres días.
Como la insulina suprime la lipólisis, es de esperar que con su administración el beneficio sea reducir el depósito adicional de lípidos en el hígado. La insulina estimula además la secreción de lípidos por perfusión hepática, pero parece necesario exponer al hígado durante largo tiempo y con altas concentraciones de insulina para lograr este efecto(18).
Glucocorticoides. Los glucocorticoides son agentes hiperglucemiantes en los rumiantes normales y cetósicos. Producen una elevación de la glicemia por cambios en la distribución y utilización de la glucosa, antes que por inducción de la NG. Esta observación se sustenta en la constatación de que durante la terapia con glucocorticoides en la oveja no se ha observado balance nitrogenado negativo, no se ha inducido el aumento de enzimas de la NG y los cambios en la cinética de la glucosa parecen indicar que la cantidad total de glucosa en el cuerpo no cambia(18).
El efecto de la menor utilización de la glucosa durante la terapia con glucocorticoides en rumiantes daría lugar a una reducción de la NG, que permite que los intermediarios del ciclo de Krebs, tales como el oxalacetato y el citrato, incrementen su concentración hacia el interior de la mitocondria, llevando como consecuencia a una reducción de la cetogénesis hepática(18).
Sin embargo varios autores afirman que los glucocorticoides tienen un escaso efecto terapéutico en la Toxemia de la gestación, más aún si se compara con los obtenidos en la cetosis bovina, sugiriendo asimismo que para obtener resultados positivos con una terapia en base a glucocorticoides, es necesario utilizar grandes dosis de corticoides de acción prolongada, ya que con las dosis usuales no se ha demostrado ningún resultado(3,32).
Rehidratación oral. En la toxemia de la gestación, además de la hipoglucemia, hay un déficit muy importante de agua y sodio; por ello se propone el uso de soluciones rehidratantes concentradas por vía oral, las cuales activan el proceso de absorción en el intestino delgado, provocando un rápido transporte de agua, glucosa y sodio. Cuando el tratamiento es aplicado en etapas iniciales de la enfermedad , la glucosa sanguínea aumenta rápidamente tras la administración de las soluciones rehidratantes conteniendo glucosa, sin embargo en períodos finales de la enfermedad la encefalopatía hipoglucémica instaurada hace irreversible la patología a pesar del tratamiento(38).
Interrupción de la gestación. La extracción del feto mediante cesárea o la inducción del parto, se fundamenta en que al frenar el drenaje de glucosa de los fetos, la madre recupera su equilibrio metabólico(11). Generalmente estos tratamientos se enfocan en salvar la madre a expensas del feto. Lo habitual es que en los rebaños no se conozca exactamente la fecha del parto, por lo tanto el neonato raramente sobrevive cuando se realiza cesárea o el parto es inducido antes de la última semana de gestación. Eliminar el feto por cesárea o provocando el parto debería reservarse para las primeras etapas de la enfermedad, antes de que la condición de la oveja sea irreversible o la muerte y descomposición fetal hayan ocurrido. La cesárea además es más apropiada en aquellos animales que hayan mostrado fallos en la respuesta a la medicación dentro de las 24 h de iniciado el tratamiento(6,7).
Para la inducción del parto se recomienda la inyección intramuscular de 10 mg de dexametasona durante 4 a 6 días entre los 133 a 135 días de la gestación, o una dosis única a partir del día 144 de la gestación, ocurriendo el parto a las 48 a 72 h posteriores al tratamiento(34). Se ha observado(6), que durante las últimas etapas de la toxemia de la gestación, la respuesta a los corticoides para inducir el parto es variable y no es fiable. Andrews(11) sugiere que esta pobre respuesta se puede relacionar con los elevados niveles de corticosteroides endógenos.
Otros tratamientos. Los receptores nucleares lípido activables o activados por proliferadores peroxisomales (PPAR) tienen un rol importante en la transcripción y regulación de los genes involucrados en el metabolismo de los lípidos, así como en la homeostasis energética(39), los cuales son asimismo activados por sustancias llamadas fibratos o ligantes(40,41). Los PPARa participan además en la homeostasis energética, regulando la expresión de genes implicados en la neoglucogénesis(40,41).
Actualmente se investiga la utilización de fibratos estimulantes de los PPARa como tratamiento de la toxemia. El ácido 2–metil–2–fenoxi–propiónico es un fibrato estimulante específico de los PPAR, actuando de la misma manera que los ligantes naturales. Al unirse a los PPAR interactuaría en la regulación de genes implicados en el metabolismo lipídico y glucídico al promover la oxidación mitocondrial y peroxisomal y la neoglucogénesis hepática(39,40), pudiéndose constituir en una nueva alternativa de tratamiento.
AGRADECIMIENTOS
Se agradece a los Dres. Edgardo Rodas de la Facultad de Veterinaria UDELAR y Felipe Prieto Montaña de la Facultad de Veterinaria de la Universidad de León, España por la corrección de esta revisión. Agradecemos asimismo a la Comisión Sectorial de Investigación Científica (CSIC), Universidad de la República, Uruguay, por su apoyo en la financiación de los trabajos de investigación.
LITERATURA CITADA
1 Harmeyer J, Schlumbohm C. Pregnancy impairs ketone body disposal in late gestating ewes: Implications for onset of pregnancy toxaemia. Res Vet Sci 2006;81(2):254–264. [ Links ]
2 West HJ. Maternal undernutrition during late pregnancy in sheep. Its relationship to maternal condition, pregnancy length, hepatic physiology and glucose metabolism. Br J Nutr 1996;75(4):593–605. [ Links ]
3 González–Montaña JR, Rejas–López J. Toxemia de la Gestación. Med Vet 1995;12(9):513–522. [ Links ]
4 Bonino J, Sienra R, Sorondo L. Enfermedades causadas por trastornos metabólicos: toxemia de la preñez. In: Bonino J, Durán del Campo A, Mari J editores. Enfermedades de los lanares II. Montevideo, Uruguay: Hemisferio Sur; 1987:239265. [ Links ]
5 Sigurdsson H. Susceptibility to pregnancy disease in ewes and its relation to gestational diabetes. Acta Vet Scand 1988b:29(3–4):407–414. [ Links ]
6 Rook JS. Pregnancy toxemia of ewes, does, and beef cows. Vet Clin North Am Food Anim Pract 2000;16(2):293–317. [ Links ]
7 Marteniuk JV, Herdt TH. Pregnancy toxemia and ketosis of ewes and does. Vet Clin North Am Food Anim Pract 1988;4(2):307–315. [ Links ]
8 Sargison N, Scott P, Penny C, Pirie R, Kelly J. Plasma enzymes and metabolites as potential prognosis indices of ovine pregnancy toxaemia. A preliminary study. Br Vet J 1994;150(3):271–276. [ Links ]
9 Cal–Pereyra L, Borteiro C, Benech A, Rodas E, Abreu MN, Cruz JC, Gonzalez–Montaña R. Histological changes of the liver and metabolic correlates in ewes with pregnancy toxemia. Arq Bras Med Vet Zootec 2009;61(2):306–312. [ Links ]
10 Chandler KD, Leury BJ, Bird, AR, Bell AW. Effects of udernutrition and exercise during late pregnancy on uterine, fetal and uteroplacental metabolism in the ewe. British J Nutrition 1985;53:625–635. [ Links ]
11 Andrews A. Pregnancy toxaemia in the ewe. In Practice 1997;19(6):306–312. [ Links ]
36 Van Saun RJ. Pregnancy toxaemia in a flock of sheep. JAVMA 2000;217(10):1536–1539. [ Links ]
12 Wastney ME, Wolff JE, Bickerstaffe R. Glucose turnover and hepatocyte glucose production of starved and Toxaemic Pregnant sheep. Aust J Biol Sci 1983;36(3);271–284. [ Links ]
13 Husted SM, Nielsen MO, Blache D, Ingsvartsen KL (2008). Glucose homeostasis and metabolic adaptation in the pregnant and lactating are affected by the level of nutrition previously provided during her late fetal live. Domestic Anim Endocrin 2008;34:419–431. [ Links ]
14 Duffield T. Subclinical ketosis in lactating dairy cattle. Vet Clin North Am Food Anim Pract 2000;16(2):231–251. [ Links ]
15 Duehelmeier R, Fluegge I, Schwert B, Parvizi N, Ganter N. Metabolic adaptation to pregnancy and lactation in German Bl ackheaded Mutton and Finn sheep ewes with different susceptibilities to pregnancy toxaemia. Small Rum Res 2011; 96:178–184. [ Links ]
16 Bell AW. Regulation of organic nutrient metabolism during transition from late pregnancy to early lactation. J Anim Sci 1995;73(9):2804–2819. [ Links ]
17 Wierda A, Verhoeff J, Van Dijk S, Dorresteijn J, Wensing T. Effects of Trembolone acetate and propileneglicol on pregnancy toxaemia in ewes. Vet Rec 1985;116(11):284–287. [ Links ]
18 H erdt TH, Emery RS. Therapy of diseases of ruminant intermediary metabolism. Vet Clin North Am Food Anim Pract 1992;8(1);91–106. [ Links ]
19 Cal–Pereyra L. Inducción experimental de Toxemia de la Gestación. Aplicación a la explotación ovina en Uruguay. 1rst ed. León, España: Universidad de León, Secretariado de Publicaciones; 2007. [ Links ]
20 González Montaña JR. Patología de la nutrición y del metabolismo. En: Fidalgo Alvarez LE, Rejas LJ, Ruiz de Gopegui R, Ramos AJJ editors. Patología Médica Veterinaria. Salamanca, Universidad de León, Universidad de Santiago de Compostela, Universidad de Zaragoza; 2003:330–379. [ Links ]
21 Kulcsár M, Dankó G, Delavaud C, Mircu C, Nikolic AJ, Gáspárdy A et al. Endocrine characteristics of late pregnant hyperketonaemic ewes and their reproductive performance following the induction of ovarian cyclicity out of breeding season. Acta Vet Hung 2006;54(2):235–249. [ Links ]
22 Jeffrey M, Higgins RJ. Brain lesions of naturally occurring pregnancy toxaemia of sheep. Vet Pathol 1992;29(4):301–307. [ Links ]
23 Contreras PA. Síndrome de movilización grasa en vacas lecheras al inicio de la lactancia y sus efectos en salud y producción de los rebaños. Arch Med Vet 1998;30(2):1–7. [ Links ]
24 Cal–Pereyra L, Benech A, Abreu MN, Borteiro C, Cruz JC, Ricciardi L, Godiño L, Nievas C, Rodas E, González–Montaña JR. Evaluación preliminar del riesgo de aparición de toxemia de la gestación en ovejas bajo diferentes manejos nutricionales y sometidas a un ayuno de 48 horas. Veterinaria (Montevideo) 2006;41(161–162):39–44. [ Links ]
25 Ford EJ, Evans J, Robinson I. Cortisol in pregnancy toxaemia of sheep. Br Vet J 1990;146(6):539–542. [ Links ]
26 Pethick DW, Lindsay DB. Metabolism of ketone bodies in pregnant sheep. Br J Nutr 1982;48(3):549–563. [ Links ]
27 Scott PR, Sargison ND, Penny CD, Strachan WD. Aqueous humours and cerebrospinal fluid collected at necropsy as indicators of ante mortem serum 3–oh butirate concentration in pregnant sheep. Br Vet J 1995;151(4):459–461. [ Links ]
28 Cantley CEL, Ford CM, Heath MF. Serum fructosamine in ovine pregnancy toxaemia: a possible prognostic index. Vet Rec 1991;128(22):525–526. [ Links ]
29 Sienra R, Bonino J, Larregui V, Echeguía M. Toxemia de la preñez II. Inducción experimental y respuesta a la terapia con glicerol – propilenglicol. Veterinaria (Montevideo) 1984;20(88–89):78–83. [ Links ]
30 Yarim GF, Ciftci G. Serum protein pattern in ewe whit pregnancy toxemia. Vet Res Commun 2009;33(5):431–438. [ Links ]
31 Yarim GR, Karahan S, Nisbet C. Elevated plasma levels of interleukin 1b, tumour necrosis factor a and monocyte chemotactic protein 1 are associated with pregnancy toxaemia in ewes. Vet Res Commun 2007;31(5):565–573. [ Links ]
32 Koenig MV, Contreras PA. Alteraciones del metabolismo energético en rumiantes y sus principales manifestaciones clínicas. Arch Med Vet 1984;16(1):7–13. [ Links ]
33 Prieto F, García PP. Los animales de granja en la investigación biomédica. En: Pérez CC, Díez I, García PP editores. Introducción a la experimentación y protección animal. Ponferrada, Universidad de León, Secretariado de Publicaciones, 1999:47–64. [ Links ]
34 Hunt ER. Treatment of pregnancy toxaemia in ewes by induction of parturition. Australian Vet J 1976;52:338–339. [ Links ]
35 Emery RS, Brown RW, Black AL. Metabolism of DL–1,2–propanediol–2–14C in a lactating cow. J Nutr 1967;92:348–352. [ Links ]
36 González MJR, Alonso DAJ, López MS, Cal PL, Prieto MF. Utilización de la glucosa vía oral para el tratamiento de la gestosis ovina. Fase preliminar. Congreso Internacional de la Federación Mediterránea de Sanidad y Producción de Rumiantes, León, España. 2001. [ Links ]
37 Brockman RP, Laarveld B. Effects of insulin on net hepatic metabolism of acetate and b–hidroxybutyrate in sheep (ovis aries). Comp Biochem Physiol A Comp Physiol 1985;81(2):255–257. [ Links ]
38 Buswell IF, Haddy IP, B ywater RJ. Treatment of pregnancy toxaemia in sheep using a concentrated oral rehydration solution. Vet Rec 1986;118:208–209. [ Links ]
39 Uauy R, Martínez J, Rojas C. Nutrición molecular, papel del sistema PPAR en el metabolismo lipídico y su importancia en obesidad y diabetes mellitus. Rev Méd Chile 2000;128(4):1–14. [ Links ]
40 De Brito–Gomes M. Glitazonas e síndrome metabólica: mecanismos de ação, fisiopatología e indicações terapéuticas. Arq Bras Endocrinol Metab 2006;50(2):271–280. [ Links ]
41 Hermes–Toros X. Farmacología do Fibratos. Arq Bras Cardiol 2005;85(5):15–16. [ Links ]

