










Serviços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista mexicana de ciencias agrícolas
versão impressa ISSN 2007-0934
Rev. Mex. Cienc. Agríc vol.7 no.4 Texcoco Mai./Jun. 2016
Articles
Optimizing a protocol for DNA isolation of leaf Saccharum officinarum
1 INIFAP-Campo Experimental Tecomán. Carretera Colima-Manzanillo km 35, Tecomán, Colima, México. (orozco.mario@inifap.gob.mx; velazquez.joaquin@inifap.gob.mx).
2 Facultad de Ciencias Biológicas y Agropecuarias-Universidad de Colima. (sguzman@ucol.mx; marcobn@me.com).
3 Instituto de Ciencias Agrícolas-Universidad Autónoma de Baja California. (michelc@uabc.edu.mx).
The DNA extraction free polysaccharides, polyphenols, plant protein and RNA is the initial step to several studies in molecular biology. There are various methods of extracting specific DNA of sugar cane (Saccharum officinarum), however, are time consuming and expensive equipment and reagents used. The aim of this study was to optimize a fast, efficient and inexpensive protocol for extracting DNA from leaves of S. officinarum. The variables evaluated were yield, purity, integrity and functionality of DNA purified from leaves of sugarcane 6 months old. Techniques spectrophotometry, agarose gel electrophoresis and molecular markers to evaluate the above variables were used. The performance variables (mg/g) and purity (A260:280 and A260:230) the extracted DNA showed highly significant differences (p˃ 0.05) protocol I. The best results were obtained with the protocol interaction I +N2, DNA yield 1.17 mg/g and 1.95 and 1.91 purities (A260:280 and A260:230). Functionality DNA by PCR amplification with the RAPD markers and TRAP bands sharp profiles generated good quality and easy to interpret. The use of the protocol I+N2 for extracting DNA from S. officinarum to obtain high yields of DNA with optimal quality and purity for applications of molecular markers is recommended.
Keywords: Saccharum officinarum; CTAB; liquid nitrogen; molecular markers
La extracción de DNA libre de polisacáridos, polifenoles, proteínas y RNA de plantas constituye el paso inicial para diversos estudios en Biología molecular. Existen diversos métodos de extracción de DNA específicos para caña de azúcar (Saccharum off icinarum), sin embargo, consumen mucho tiempo y utilizan reactivos y equipos costosos. El objetivo del presente estudio fue optimizar un protocolo rápido, eficiente y debajo costo para extraer DNA de hojas de S. officinarum. Las variables evaluadas fueron rendimiento, pureza, integridad y funcionalidad del DNA purificado de hojas de caña de azúcar de 6 meses de edad. Se utilizaron técnicas de espectrofotometría, electroforesis en geles de agarosa y marcadores moleculares para evaluar las variables anteriores. Las variables rendimiento (mg/g) y pureza (A260:280 y A260:230) del DNA extraído mostraron diferencias altamente significativas (p˃ 0.05) con el protocolo I. Los mejores resultados se obtuvieron con la interacción del protocolo I +N2, con un rendimiento de DNA de 1.17 mg/g y purezas de 1.95 y 1.91 (A260:280 y A260:230). La funcionalidad del DNA por amplificación por PCR con los marcadores RAPD y TRAP generaron perfiles de bandas nítidas de buena calidad y de fácil interpretación. Se recomienda el uso del protocolo I +N2 para la extracción de DNA de S. officinarum para obtener elevados rendimientos de DNA con calidad y pureza óptima para aplicaciones de marcadores moleculares.
Palabras clave: Saccharum officinarum; nitrógeno líquido; CTAB; marcadores moleculares
Introduction
The extraction protocols and nucleic acid purification are essential for most applications in molecular biology. Techniques such as real-time PCR, microarrays and molecular markers using DNA as a starting point (Menossi et al., 2007; Gupta et al., 2010; Khan et al., 2011; Chang et al., 2012; Devarumath et al., 2013; Garces et al., 2014). In plants, the success in various biotechnological applications involving the use of DNA depends largely on the yield and purity obtained. Most of existing protocols, to isolate and purify DNA of good quality, are based on the method of cetyltrimethylammonium bromide (CTAB), being economical (Murray and Thompson, 1980; Doyle and Doyle, 1990; Porebski et al., 1997; Hossain et al., 2006; Sahu et al., 2012).
The amount and types of secondary metabolites that secrete plants (flavonoids, terpenes, polyphenols, quinones and alkaloids) are varied among species, therefore it is not possible to apply a method of universal extraction (Khanuja et al., 1999). Often researchers modified a protocol for purification of DNA for a given species. Saccharum officinarum contains high levels of polysaccharides, polyphenols, RNA and proteins, which are a source of contamination to obtain pure DNA that can be used in subsequent enzymatic reactions (Honeycutt et al., 1992; Aljanabi et al., 1999; Hossain et al., 2006). Honeycutt et al. (1992) developed a method for extraction of DNA species of Saccharum, between 0.07 to 0.28 mg reporting yields of DNA per gram of fresh tissue and purity of 1.7 (A260:280).
The DNA was used in PCR-based methodologies, restriction enzymes, Southern hybridization and sequencing. Moreover, Aljanabi et al. (1999), a protocol optimized to isolate large amounts of DNA (0.5-0.8 mg/g) and purity between 1.76-1.96 (A260:280) from sugar cane meristems and without using N2 to grind tissue. The DNA obtained was feasible for use with restriction enzymes, PCR and RFLP. While Hossain et al. (2006) compared three methods of extracting DNA making some modifications to the methodology described by Aljanabi et al. (1999).
More recently, Vaze et al. (2010) developed a specific protocol to isolate genomic DNA from leaf tissue dry sugar cane without the use of N2, performance reporting DNA was 0.25-1.00 mg/g purity 1.7-1.8 (A260:280) ; This DNA was successfully used in RAPD and ISSR markers for genetic diversity studies. Often commercial kits are used to extract DNA from plants, including sugar cane, however, it is widely reported that although significantly reduce time, prove to be very costly compared to the conventional methods, plus yields are obtained very low DNA (Castillo-Reyes et al., 2004; Niu et al., 2008; Chen et al., 2010; Perez-Almeida et al., 2011; Sahu et al., 2012; Huaqiang et al., 2013a).
Although there are reports about specific protocols DNA extraction for sugarcane, most time consuming and require reagents and expensive equipment (Honeycutt et al.,1992; Aljanabi et al., 1999; Hossain et al., 2006; Vaze et al., 2010). The aim of this study was to establish an optimized protocol for purifying DNA from S. officinarum leaves at high concentrations and optimal purity, quickly and economically. With the results obtained have a standardized to purify DNA from S. officinarum that can be used in molecular markers in breeding programs protocol.
Materials and methods
Two extraction protocols and DNA purification based on the method of CTAB were established. The I protocol was based as described by Doyle and Doyle (1990) with some modifications included using proteinase K and polyvinylpyrrolidone (PVP) in homogenization buffer, an extraction with phenol-chloroform and the use of R Nase A in the final resuspension of the DNA in TE buffer. Protocol II derived from the work described by Aljanabi et al. (1999) unchanged. The tests were conducted in the laboratory of Plant Biotechnology Experimental Field-INIFAP Tecoman, Colima, Mexico.
Vegetal material. The young healthy leaves and 6 months old plants sugar cane field were harvested established. Leaf samples were transported to the laboratory refrigerated and stored at -70 °C until use. For all samples DNA was extracted from 100 mg leaf tissue. Grinding tissue +N2 was performed in mortar using a pistil; and the required amount of tissue recovered with a spoon; while for grinding -N2 the homogenization buffer he was added to the tissue in the mortar and mixed with micropipette recovered.
Protocol I. The tissue was macerated (+N2 and -N2) and homogenized with 800 μL of CTAB buffer [100 mM of Tris-HClpH8, 50m M of EDTA pH8, 1.4 MofNaCl,3%(p/v) of CTAB and 1% (p/v) of PVP]. Subsequently was added concentrated 10 μL of 2-β-mercaptoethanol and 5 μL of proteinase K (20 mg/mL). Samples incubated for 60 min at 65 °C in a water bath and mixed by inversion every 5 minutes. The chloroform: phenol 800 μL of the solution was added isoamyl alcohol in a ratio of 25:24:1 was vigorously mixed using a vortex and centrifuged at 14 000 rpm at 4 °C for 10 min.
The recovered supernatant was placed in 1.5 mL tubes and the DNA was precipitated with 0.6 volumes of cold isopropanol and 0.1 volume of 3 M sodium acetate pH 5.2 for 30 min at -20 °C. The mixture was centrifuged at 14 000 rpm at 4 °C for 10 min. The supernatant was decanted and 500 μL was added to 70% ethanol; was manually stirred until detaching the DNA pellet and the tubes at 14 000 rpm at 4 °C for 5 min centrifuged, the aqueous phase was decanted and inverted onto absorbent paper to remove excess ethanol. The DNA pellet was dried at room temperature and resuspended in 40 μL of buffer (10 mM of Tris-HCl, 1 mM of EDTA pH 8) TE. Finally 1 μL of RNase A (10 mg/mL) was added, mixed with a micropipette, incubated at room temperature for 5 minutes and stored at -20 °C.
Protocol II. The tissue (+N2 and -N2) macerated and mixed with 500 mL of homogenization buffer [200 mM of Tris- HCl pH 8, 50 mM of EDTA pH 8, 2.2 M of NaCl, 2% (p/v) of CTAB, 0.06% (p/v) of sodium sulfite]. The 100 μL of N-lauryl sarcosine 5% (p/v), 100 μL of 10% PVP (p/v) and 100 μL of CTAB 20% (p/v) was added. It was incubated at 65 °C for 60 min in a water bath and mixed by inversion every 5 minutes. The samples were cooled to room temperature and a volume of the phenol solution was added: chloroform: isoamyl alcohol (25:24:1). Was mixed by inversion and centrifuged at 14 000 rpm for 10 min at 4 °C. the aqueous phase was recovered and transferred to sterile tubes. A volume of cold isopropanol followed of 100 μL 6 M NaCl and incubated at -20 °C for 60 min was added. Subsequently, the tubes were centrifuged for 10 min at 14 000 rpm. The DNA pellet was washed with 1 mL of 70% ethanol and dried at room temperature. DNA was resuspended in 40 mL of buffer (10 mM Tris-HCl, 1mM EDTA, pH 8) TE and stored at -20 °C.
Quantitation, purity and integrity of the DNA. The DNA concentration (ng/μL) was quantitated with a spectrophotometer NanoDrop Thermo Scientific and converted to mg/g. As a criterion of purity A260:280 and A260:230 relations were considered. The integrity of nucleic acids was verified by separation by gel electrophoresis 1% agarose 1 μg genomic DNA, stained with ethidium bromide (10 mg/mL) and visualized with UV light in a foto documentador.
Purified DNA functionality
RAPD marker. Oligonucleotide was used OPA-10-5′- GTGATCGCAG-3’ (Pan et al., 1996). The volume of the PCR reaction was 25 μL containing 1 X reaction buffer, 1 X, 1.5 mM de MgCl2, 1 μM of each oligonucleotide, 100 μM of dNTPs, 1.25 U of DNA polymerase (BioTecMol) and 50 ng of genomic DNA. Amplification conditions for the reaction were carried were based according to Chen et al, 2011, with some modifications. Initial denaturation at 94 °C-4 min, followed by 45 cycles of 94 °C-1 min, 36 °C-1 min and 72 °C-2 min denaturation, alignment and extension, respectively, and a final extension at 72 °C-7 min. The PCR products were separated by horizontal electrophoresis in agarose gels 1.5% (p/v) with TAE buffer stained with ethidium bromide (10 mg/mL).
TRAP marker. The game was used oligonucleotide Arbi 3 5 ́-GACTGCGTACGAATTTGA-3 ́ and SuSy -5′-GGAGGAGCTGAGTGTTTC-3’ (Hu and Vick, 2003; Devarumath et al., 2013). The PCR reaction was performed in a volume of 25 μL containing reaction buffer 1 X, 1.5 mM de MgCl2, 1 μM of each oligonucleotide, 100 μM of dNTPs, 1.25 U of DNA polymerase (BioTecMol) and 50 ng of DNA genomic. Amplification conditions for the reaction were as follows: initial denaturation at 94 °C-2 min, followed by 5 cycles of 94 °C-45 s, 35 °C-45 s and 72 °C-1 min denaturation, alignment and extension, respectively; followed by 35 cycles of 94 °C-45 s, 50 °C-45 s and 72 °C-1 min denaturation, alignment and extension, respectively. Final extension was 72 °C-7 min (Hu and Vick, 2003). The PCR products were separated by horizontal electrophoresis in agarose gels 1.5% (p/v) with TAE buffer and stained with ethidium bromide (10 mg/mL).
Statistical analysis. The yield (mg/g) and purity (A260:280and A260:230) were analyzed for DNA analysis of variance using a completely randomized design factorial 2 (protocols) x 2 ((+N2 and -N2) with, giving a total of 4 treatments and 10 repetitions. Experimental unit was considered as a sample of DNA from sugar cane. Test Duncan's multiple range was used to compare mean values between variables than 95% probability using the SAS statistical package. The integrity and functionality of DNA were visually assessed with agarose gels.
Results and discussion
DNA yield
The effect of using N2 performance on DNA obtained from leaves of sugarcane shown in Table 1. The average values of extracted DNA yield +N2 (0.68 mg/g), were significantly higher than those obtained without the use of the same (0.50 mg/g) (p˃ 0.05). These results exceed those reported by other authors, who extracted DNA from sugar cane, without the use of N2 and with yields of DNA below 0.30 mg/g of leaf tissue (Honeycutt et al., 1992; Hossain et al., 2006; Vaze et al., 2010).
Table1 Performance DNA extracted from young leaves of S. officinarum (+N2= with liquid nitrogen; -N2= no liquid nitrogen).
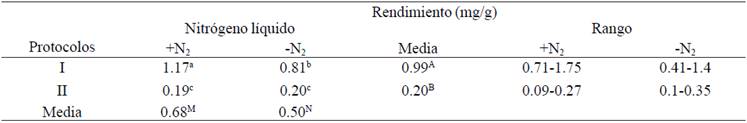
The results obtained in this study indicate that the use of N2 increased performance S. officinarum DNA. Generally, the use of N2 is widely documented in plants (Ahmad et al., 2004; Niu et al., 2008; Arif et al.,2010; Sharma et al., 2010; Cingilli-Vural y Dageri, 2011; Doosty et al., 2012; Huaqiang et al., 2013a; Huaqiang et al., 2013b); fungi (Niu et al., 2008; Motkova y Vytrasova, 2011; Caceres et al., 2012; Prabha et al., 2013) and animals (Gross-Bellard et al., 1973; Chen et al., 2010; Eschbach, 2012). Using N2 freezes -196 to -210 °C tissue samples, facilitating all ruptured cell walls and membranes, allowing exposure of nucleic acids with extraction buffer (Caceres et al., 2012).
This suggests that the use of N2 probably releases a larger amount of nucleic acids of a larger number of cells, yielding improved performance. Also reported obtaining DNA of 2012). These results suggest that the use of spermidine and PEG is not necessary for DNA quality S. officinarum, reducing the cost of the extraction process.
Moreover, Aljanabi et al. (1999), obtained high yields of DNA from 0.50 to 0.80 mg/g tissue meristem sugarcane using the "Ultra-Turax" equipment for homogenization of samples and a modification of the CTAB method of Doyle and Doyle (1990) which included high concentrations of CTAB (20%) and NaCl (6 M) to remove polysaccharides. In this paper the methodology described by the aforementioned author (Protocol II), using 100 mg of leaf tissue (not meristems) was reproduced. The DNA yields obtained were very low and remained in a range of 0.10 to 0.30 mg/g. These results indicate that the type of fabric used and equipment for the extraction of DNA from S. officinarum could explain the differences between the yields obtained with the two protocols. Therefore, it is not necessary to use high concentrations of CTAB and NaCl to obtain high purity DNA from young leaves of S. officinarum.
Hossain et al. (2006) evaluated three DNA extraction protocols for sugar cane from leaves and meristems, with a change in the methodology proposed by Aljanabi et al. (1999). The authors replaced the N-lauryl sarcosine sodium dodecyl sulfate by (SDS), both anionic detergents which break the covalent bonds between proteins, denaturing the DNA purification process. The authors obtained a yield of DNA of 0.04 mg/g, significantly lower the here reported for leaves (0.20 mg/g) (Table 1) using the protocol II values. The use of different anionic detergents may have different effectiveness on the breaking of covalent bonds of proteins, so probably the use of N-lauryl sarcosine could increase yields DNA of S. officinarum protocol II. Moreover, Vaze et al. (2010) reported low yields DNA 0.02-0.10 mg/g dry leaf tissue of sugarcane.
These variations between yields can be explained by the type and condition of tissue used, as in the present study used young leaves, which are in constant cell division, so there are as many cells per gram of tissue; addition, the concentration of phenolic compounds, polysaccharides and secondary metabolites is lower (Jobes et al., 1995). Honeycutt et al. (1992) used mature leaves, where the concentration of phenolic compounds, polysaccharides and secondary metabolites is increased and the number of cells per gram of tissue is less compared to young leaves. Moreover, Aljanabi et al. (1999) included sugar cane meristems, which have similar properties to the young leaves. Meanwhile, Hossain et al. (2006) made use of meristems and leaves; while Vaze et al. (2010) extracted the DNA from dried leaves of cane sugar cane, where the yield and quality of DNA are low and can be degraded. The above results suggest the use of young leaves of S. officinarum extract and purify DNA for high throughput and acceptable purity.
Purity and integrity of DNA
The effect of the use of N2 and DNA extraction protocols on purity (A260:A280 and A260:A230) of the DNA obtained from sugar cane leaves shown in Table 2 and 3. A pure sample of DNA has a ratio A260:A280 between 1.8-1.9, while the A260:A230 should be 1.8-2.2 (Gallagher and Desjardins, 2011). The purities (A260:A280) +N2 and -N2 were 2.01 and 2.00, respectively, so no significant difference with this variable (p˃ 0.05) were detected. Moreover, highly significant differences were detected using protocols (p˃ 0.05) (Table 2), while the protocols *N2 interaction was significant (p˃ 0.05). These results are consistent with those reported in sugar cane Aljanabi et al. (1999) and Vaze et al. (2010), while Honeycutt et al. (1992) and Hossain et al. (2006) reported purities below 1.8, so that DNA is probably slightly contaminated by proteins and/or phenols (Gallagher and Desjardins, 2011; Huaqiang et al., 2013a).
Table 2 Comparison of purity (A260:A280 nm) of DNA extracted from S. officinarum (+N2= with liquid nitrogen; -N2= no liquid nitrogen).
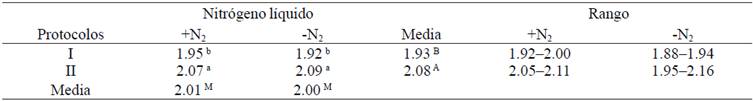
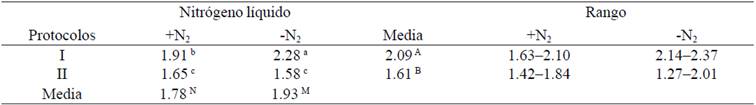
Table 3 Comparison of purity (A260:A230 nm) of DNA extracted from S. officinarum (+N2= with liquid nitrogen; -N2= no liquid nitrogen).
Aljanabi et al. (1999) obtained for all samples A260/280 of between 1.76-1.96 and reported that the RNA was degraded in the extraction process, however, in this study using the same methodology (Protocol II), was reported for all samples values of the ratio A260/280 above 2 (Table 2), indicating the presence of RNA, which was not degraded during the extraction process as described by Aljanabi et al. (1999), so DNA quantifications II protocol could be overestimated. This was confirmed by electrophoresis on an agarose gel (Figure 1, A). These results suggest the use of RNase A in the DNA extraction process. Using the protocol I RNase A was not eliminated, this suggests that it may be possible omission of incubation at 37 °C and subsequent deactivation and cleaning of this enzyme, saving time. It has not been observed that there is inhibition effect of RNase A in the PCR reactions, so the inclusion of RNase A (10 mg/mL) in the final resuspension of DNA is recommended.

Figure 1 Gel electrophoresis in agarose genomic DNA of S. officinarum extracted with two protocols based on the CTAB method from leaf tissue. He was loaded in each lane 1 μg of DNA. MPM= molecular weight marker, 1 Kb Plus (Invitrogen)A) +N2 extraction. Protocol I= lanes 1-9; protocol II= lanes 10-18. B) Removal -N2. Protocol I= lanes 1-4; protocol II= lanes 5-8.
The purities (A260:A230) +N2 and -N2 being 1.78 and 1.93 were respectively detected with this variable significant differences (p˃ 0.05), while the effect of the protocols was highly significant (p˃ 0.05) and interaction protocols *N2 was significant (Table 3). Data reported to the protocol I indicated that the DNA is free polysaccharides. The DNA extraction methods specific for S. officinarum described by Honeycutt et al. (1992), Aljanabi et al. (1999), Hossain et al. (2006) and Vaze et al. (2010) do not report the ratio A260:A230 as second criterion of purity of DNA, to which the polysaccharides are detected. In the protocol II showed lower values of A260:A230 of the 1.8 (Table 3), so probably the DNA are slightly contaminated with polysaccharides or phenols. I use +N2 protocol is recommended to extract DNA from leaves of S. officinarum as both criteria of purity (A260:A280 and A260:A230) they were optimal.
The integrity of genomic DNA influenced by the use of N2 and the two extraction protocols in young leaves of sugarcane in Figure 1. Agarose gels with DNA extracted from leaves were ground with N2 showed banding patterns genomic DNA of high molecular weight in all lanes with two protocols evaluated (Figure 1-A), so that no degradation of the genomic DNA was detected. The protocol II samples (Lanes 10-18) RNA showed contamination due to the omission of treatment with RNase A as described by Aljanabi et al. (1999). The protocol I turned out to be the best for full DNA without RNA residues (Figure 1-A, lanes 1-9). The genomic DNA obtained is similar integrity to that reported by Aljanabi et al. (1999) and Vaze et al. (2010) in sugarcane and other authors who used the method of CTAB in cowpea (Huaqiang et al., 2013a; Huaqiang et al., 2013b). Moreover, the DNA extracted DNA patterns showed -N2 scanning using the two protocols (Figure 1-B), indicating degradation. Similar results were obtained by Honeycutt et al. (1992). The use of N2 is recommended to extract DNA with acceptable integrity S. officinarum leaves.
Functionality
In the Figure 2 shows the profiles of two molecular markers used to verify the performance of isolated DNA protocols set out in this work. The RAPD marker for all samples amplified perfectly without any effect by the variables N2 and protocols (Figure 2-A) is noticed. Moreover, waste of RNA (Figure 1, A) protocol II had no negative effect on the PCR reaction, a good profile of amplified bands. Moreover, with the marker pattern TRAP bands with better quality samples they were treated +N2 was obtained (Figure 2-B, lanes 1 and 2). The extracted samples -N2 showed a pattern of diffuse bands and poor quality for interpretation (Figure 2-B, lanes 4 and 5). It is likely to raise the profile of bands with changing the annealing temperature (Tm) of the oligonucleotide. Moreover, considering that in RAPD markers is complicated reproducibility in other labs, it is important that the DNA be optimal values of A 260:280 and A260:230 to over come this problem; so the use of the protocol I +N2 is recommended to extract DNA from S. officinarum sheets for use with molecular markers.
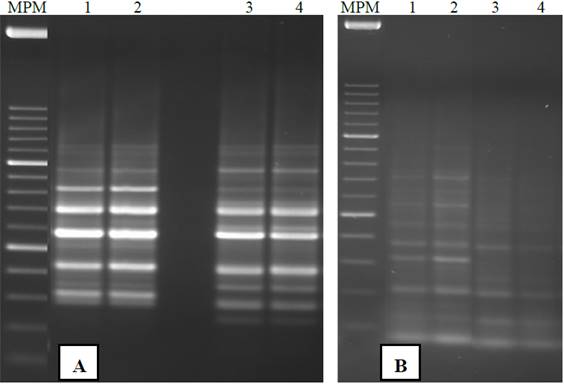
Figure 2 Molecular markers with the DNA of S. officinarum leaves extracted with two protocols based on the CTAB method. MPM= molecular weight marker 100 bp (Invitrogen). A) RAPD with oligonucleotide OPA 10. B) TRAP with oligonucleotide SuSy and Arbi 3. Lanes 1 and 2= ground +N2 protocols I and II, respectively samples. Lanes 4 and 5= ground samples -N2 protocols I and II, respectively.
In addition to the molecular markers used to explore the genetic diversity of S. officinarum, the DNA obtained with I +N2 protocol has been successfully used in PCR-based methodologies and sequencing for detection of pathogens in sugar cane: Xanthomonas albilineans, Leifsonia xyli subsp. xyli, Puccinia melanocephala and P. kuehnii cause diseases of economic importance: scald, rickets, rust brown and orange, respectively rust. It has also been used in real time PCR for detection of Candidatus Liberibacter asiaticus, causative agent of Huanglongbing in several species of citrus (Citrus aurantifolia, C. limon y citranges) (data not shown).
Conclusions
Modifications to the CTAB for DNA extraction method of S. officinarum allowed optimizing a protocol for DNA quality and high quantity. Highest yields were obtained with DNA interaction protocol I +N2 for grinding leaf tissue. Furthermore, this same protocol consumed the least amount of time.
The purity (A260:280 and A260:230) isolated DNA was higher quality with the use of the protocol I +N2. The DNA integrity presented no degradation +N2, while degradation occurred in DNA samples extracted with the two protocols -N evaluated.
As for the functionality of DNA profiles sharp and well marked bands were obtained with the use of RAPD molecular marker +N2 and -N2. While for a better marker TRAP banding pattern with the inclusion of N in either protocol it was achieved. It is recommended to use I+N2 protocol for extracting DNA from sugar cane leaves that could be used with various molecular markers in breeding programs.
Literatura citada
Ahmad, S. M.; Ganaie, M. M.; Qazi, P. H.; Verma, V.; Basir, S. F. and Qazi, G. N. 2004. Rapid DNA isolation protocol for angiospermic plants. Bulgarian J. Plant Physiol. 30:25-33. [ Links ]
Aljanabi, S. M.; Forget, L. and Dookun, A. 1999. An improved rapid protocol for the isolation of polysaccharide and polyphenol-free sugarcane DNA. Plant Mol. Biol. Reporter. 17(3):1-8. [ Links ]
Arif, I. A.; Bakir, M. A.; Khan, H. A.; Ahamed, A.; Al Farhan, A. H.; Al Homaidan, A. A.; Al Sadoon, M.; Bahkali, A. H. and Shobrak, M. 2010. A simple method for DNA extraction from mature date palm leaves: impact of sand grinding and composition of lysis buffer. Int. J. Mol. Sci. 11(9):3149-3157. [ Links ]
Cáceres, P.; Cordero, C.; González, G.; Quiroz, K.; Bobadilla, J. C.; Bravo, C.; Caligari, P. D. S.; Carrasco, B. and García-González, R. 2012. Efficient protocols for the extraction of microbial DNA from the rhizosphere of hydrophilic forests in Chile. Ciencia e Investigación Agraria. 39(3):585-592. [ Links ]
Castillo-Reyes, F.; García-Villanueva, A. P.; Gómez-Martínez, M. y Reyes-Valdés, M. H. 2004. Comparación de tres métodos para el aislamiento de ADN en girasol. Rev. Agraria. 52(1):24-28. [ Links ]
Chang, D.; Yang, F. Y.; Yan, J. J.; Wu, Y. Q.; Bai, S. Q.; Liang, X. Z.; Zhang, Y. W. and Gan, Y. M. 2012. SRAP analysis of genetic diversity of nine native populations of wild sugarcane, Saccharum spontaneum, from Sichuan, China. Genetics Mol. Res. 11(2):1245-1253. [ Links ]
Chen, H.; Rangasamy, M.; Tan, S. Y.; Wang, H. and Siegfried, B. D. 2010. Evaluation of five methods for total DNA extraction from western corn rootworm beetles. PLoS ONE. 5(8):1-6. [ Links ]
Chen, P. H.; Salazar, E.; Fernández, H.; Castro, L.; Russo, A. y Vázquez, S. 2011. Detección de polimorfismos RAPD en materiales de Musa sp. con respuesta diferencial al ataque de Xanthomonas campestris pv. musacearum. Agronomía Tropical. 61(2):125-132. [ Links ]
Cingilli-Vural, H. and Dageri, N. 2011. Evaluation of efficiency of different DNA extraction protocols and molecular identification of commercial soybean (Glycine max., L.) growing in Turkey. J. Appl. Biol. Sci. 5(2):12-19. [ Links ]
Devarumath, R. M.; Kalwade, S. B.; Bundock, P.; Eliott, F. G. and Henry, R. 2013. Independent target region amplification polymorphism and single-nucleotide polymorphism marker utility in genetic evaluation of sugarcane genotypes. Plant Breed. 132(6):736-747. [ Links ]
Doosty, B.; Drikvand, R.; Salahvarzi, E.; Amiri, H. and Hadian, J. 2012. Comparative analysis and optimization of different DNA extraction protocols in Satureja khuzistanica. Int. J. Biol. 4(4):111-116. [ Links ]
Doyle, J. J. and Doyle, J. L. 1990. Isolation of plant DNA from fresh tissue. Focus. 12:13-15. [ Links ]
Eschbach, E. 2012. Ascertaining optimal protocols for DNA extraction of different qualities of pike (Esox lucius) tissue samples a comparison of commonly used solid phase extraction methods. Environ. Biotechnol. 8:7-14. [ Links ]
Gallagher, S. R. and Desjardins, P. 2011. Quantitation of nucleic acids and proteins. Current Protocols Essential Laboratory Techniques. 5:1-36. [ Links ]
Garces, F. F.; Gutierrez, A. and Hoy, J. W. 2014. Detection and quantification of Xhantomonas albilineans by qPCR and potencial characterization of sugarcane resistance to leaf scald. Plant Dis. 98(1):121-126. [ Links ]
Gross-Bellard, M.; Oudet, P. and Chambon, P. 1973. Isolation of high-molecular-weight DNA from mammalian cells. Eur. J. Biochem. 36(1):32-38. [ Links ]
Gupta, V. K.; Misra, A. K.; Gupta, A.; Pandey, B. K. and Gaur, R. K. 2010. RAPD-PCR of Trichoderma isolates and in vitro antagonism against Fusarium wilt pathogens of Psidium guajava L. J. Plant Protec. Res. 50(3):256-262. [ Links ]
Honeycutt, R. J.; Sobral, B. W. S.; Keim, P. and Irvine, J. E. 1992. A rapid DNA extraction method for sugarcane and its relatives. Plant Mol. Biol. Reporter. 10(1):66-72. [ Links ]
Hossain, M. A.; Shaik, M. M.; Shahnawaz, R. M. S.; Islam, N. and Miah, M.A.S. 2006. Quality DNA isolation using different methods of sugarcane (Saccharum officinarum L.). Bangladesh J. Sugarcane. 28:65-69. [ Links ]
Hu, J. and Vick, B. 2003. Target region amplification, polymorphism: a novel marker technique for plant genotypes. Plant Mol. Biol. Reporter. 20(3):289-294. [ Links ]
Huaqiang, T.; Haitao, H.; Mamman, T.; Jianyao, M. and Huanxiu, L. 2013a. Comparative analysis of six DNA extraction methods in cowpea (Vigna unguiculata L. Walp). J. Agric. Sci. 5(7):82-90. [ Links ]
Huaqiang, T.; Manman, T.; Li, Z.; Yanxia, Z. and Huanxiu, L. 2013b. The effects of three different grinding methods in DNA extraction of cowpea (Vigna unguiculata L. Walp). Afr. J. Biotechnol. 12(16):1946-1951. [ Links ]
Jobes, D. V.; Hurley, D. L. and Thien, L. B. 1995. Plant DNA isolation: a method to efficiently remove polyphenolics, polysaccharides, and RNA. Taxon. 44:349-386. [ Links ]
Khan, I. A.; Bibi, S.; Yasmeen, S.; Seema, N.; Khatri, A.; Siddiqui, M. A.; Nizamani, G. S. and Afghan, S. 2011. Identification of elite sugarcane clones through TRAP. Pak. J. Bot. 43(1):261-269. [ Links ]
Khanuja, S. P. S.; Shasany, A. K.; Darokar, M. P. and Kumar, S. 1999. Rapid isolation of DNA from dry and fresh samples of plants producing large amounts of secondary metabolites and essential oils. Plant Mol. Biol. Reporter. 17(1):1-7. [ Links ]
Menossi, M.; Silva-Filho, M. C.; Vincentz, M.; Van-Sluys, M. A. and Souza, G. M. 2007. Sugarcane functional genomics: gene discovery for agronomic trait development. Int. J. Plant Gen. 2008:1-11. [ Links ]
Motkova, P. and Vytrasová, J. 2011. Comparison of methods for isolating fungal DNA. Czech J. Food Sci. 29(S):76-85. [ Links ]
Murray, M. G. and Thompson, W. F. 1980. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Research. 8(19):4321-4325. [ Links ]
Niu, C.; Kebede, H.; Auld, D. K.; Woodward, J. E.; Burow, G. and Wright, R. J. 2008. A safe inexpensive method to isolate high quality plant and fungal DNA in an open laboratory environment. Afr. J. Biotechnol. 7(16):2818-2822. [ Links ]
Pan, Y. B.; Burner, D. M.; Ehrlich, K. C.; Grisham, M. P. and Wei, Q. 1996. Analysis of primer-derived, nonspecific amplification products in RAPD-PCR. BioTechniques. 22(6):1071-1077. [ Links ]
Paithankar, K. R. and Prasad, K. S. N. 1991. Precipitation of DNA by polyethylene glycol and ethanol. Nucleic Acids Research. 19(6):1346. [ Links ]
Pérez-Almeida, I.; Graterol, L. A.; Osorio, G.; Ramis, C.; Bedoya, A. M.; Figueroa-Ruiz, R.; Molina, S. and Infante, D. 2011. Métodos modificados de obtención de ADN genómico en orquídeas (Cattleya spp.) para amplificación con marcadores moleculares. Bioagro. 23(1):27-34. [ Links ]
Porebski, S.; Bailey, L. G. and Baum, B. R. 1997. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Reporter. 15(1):8-15. [ Links ]
Prabha, T. R.; Revathi, K.; Vinod, M. S.; Shanthakumar, S. P. and Bernard, P. 2013. A simple method for total genomic DNA extraction from water moulds. Plant Breed. Gen. Res. 104(30):585-592. [ Links ]
Sahu, S. K.; Thangaraj, M. and Kathiresan, K. 2012. DNA extraction protocol for plants with high levels of secondary metabolites and polysaccharides without using liquid nitrogen and phenol. Int. Scholarly Res. Network Mol. Biol. 1:1-6. [ Links ]
Sambrook, J.; Russell, D. W. and Sambrook, J. 2001. Molecular cloning. Cold Spring Harbor Laboratory 3rd ed. NY, USA. 2100 p. [ Links ]
Sharma, P.; Joshi, N. and Sharma, A. 2010. Isolation of genomic DNA from medicinal plants without liquid nitrogen. Ind. J. Exp. Biol. 48(6):610-614. [ Links ]
Shahriar, M.; Haque, M. R.; Kabir, S.; Dewan, I. and Bhuyian, M. A. 2011. Effect of proteinase-K on genomic DNA extraction from Gram-positive strains. Stamford J. Pharm. Sci. 4(1):53-57. [ Links ]
Takakura, K. and Nishio, T. 2012. Safer DNA extraction from plant tissues using sucrose buffer and glass fiber filter. J. Plant Res. 125(6):805-807. [ Links ]
Tung-Nguyen, C. T.; Son, R.; Raha, A. R.; Lai, O. M. and Clemente- Michael, W. V. L. 2009. Comparison of DNA extraction efficiencies using various methods for the detection of genetically modified organisms (GMOs). Int. Food Res. J. 16(1):21-30. [ Links ]
Vaze, A.; Nerkar, G.; Pagariya, M.; Devarumath, R. M. and Prasad, D. T. 2010. Isolation and PCR amplification of genomic DNA from dry leaf samples of sugarcane. Int. J. Pharma Bio Sci. 1(2):1-6. [ Links ]
Wallace, H. M.; Fraser, A. V. and Hughes, A. 2003. A perspective of polyamine metabolism. Bioch. J. 376(1):1-14. [ Links ]
Received: March 2016; Accepted: June 2016
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
