











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Los protocolos de extracción y purificación de ácidos nucleicos son esenciales para la mayoría de las aplicaciones en biología molecular. Técnicas como PCR tiempo real, microarreglos y marcadores moleculares utilizan DNA como punto de partida (Menossi et al., 2007; Gupta et al., 2010; Khan et al., 2011; Chang et al., 2012; Devarumath et al., 2013; Garces et al., 2014). En plantas, el éxito en las diversas aplicaciones biotecnológicas que involucran el uso de DNA depende en gran medida del rendimiento y pureza que se obtiene. La mayoría de los protocolos existentes, para aislar y purificar DNA de buena calidad, están basados en el método del bromuro de cetiltrimetilamonio (CTAB), por ser económico (Murray y Thompson, 1980; Doyle y Doyle, 1990; Porebski et al., 1997; Hossain et al., 2006; Sahu et al., 2012).
La cantidad y tipos de metabolitos secundarios que secretan las plantas (flavonoides, terpenos, polifenoles, quinonas y alcaloides), son muy variados entre especies, por lo tanto, no es posible aplicar un método de extracción universal (Khanuja et al., 1999). Es frecuente que los investigadores modifiquen un protocolo de purificación de DNA para una especie determinada. Saccharum officinarum contiene altos niveles de polisacáridos, polifenoles, RNA y proteínas, que constituyen una fuente de contaminación para obtener DNA puro que pueda ser usado en subsecuentes reacciones enzimáticas (Honeycutt et al., 1992; Aljanabi et al., 1999; Hossain et al., 2006). Honeycutt et al. (1992), desarrollaron un método de extracción de DNA para especies del género Saccharum, reportando rendimientos entre 0.07-0.28 mg de DNA por gramo de tejido fresco y pureza de 1.7 (A260:280).
El DNA pudo emplearse en metodologías basadas en PCR, enzimas de restricción, hibridación Southern y secuenciación. Por otra parte, Aljanabi et al. (1999), optimizaron un protocolo para aislar grandes cantidades de DNA (0.5-0.8 mg/g) y de pureza entre 1.76-1.96 (A260:280) a partir de meristemos de caña de azúcar y sin hacer uso de N2 para moler el tejido. El DNA obtenido fue viable para ser utilizado con enzimas de restricción, PCR y RFLP. En tanto que Hossain et al. (2006), compararon tres métodos de extracción de DNA haciendo algunas modificaciones a la metodología descrita por Aljanabi et al. (1999).
Más recientemente, Vaze et al. (2010), desarrollaron un protocolo específico para aislar DNA genómico de tejido foliar seco de caña de azúcar sin el uso de N2, el rendimiento que reportaron de DNA fue de 0.25-1.00mg/g y pureza de1.7- 1.8 (A260:280); este DNAse utilizó exitosamente en marcadores RAPD e ISSR para estudios de diversidad genética. Frecuentemente se utilizan kits comerciales para extraer DNA de plantas, incluyendo caña de azúcar, sin embargo, está ampliamente reportado que a pesar de que reducen significativamente los tiempos, resultan ser muy costosos en comparación con los métodos clásicos, además de que se obtienen rendimientos muy bajos de DNA (Castillo-Reyes et al., 2004; Niu et al., 2008; Chen et al., 2010; Pérez-Almeida et al., 2011; Sahu et al., 2012; Huaqiang et al., 2013a).
A pesar de que existen reportes sobre protocolos específicos de extracción de DNA para caña de azúcar, la mayoría consumen mucho tiempo y requieren reactivos y equipos costosos (Honeycutt et al., 1992; Aljanabi et al., 1999; Hossain et al., 2006; Vaze et al., 2010). El objetivo del presente estudio fue establecer un protocolo optimizado para purificar DNA de hojas de S.officinaruma concentraciones altas y pureza óptima, de forma rápida y económica. Con los resultados obtenidos se dispondrá de un protocolo estandarizado para purificar DNA de S. officinarum que pueda ser utilizado en marcadores moleculares en programas de mejoramiento genético.
Materiales y métodos
Se establecieron dos protocolos de extracción y purificación de DNA basados en el método de CTAB. El protocolo I se fundamentó según lo descrito por Doyle y Doyle (1990) con algunas modificaciones que incluyeron el uso de proteínasa K y polivinilpirrolidona (PVP) en el buffer de homogeneización, una extracción con fenol-cloroformo y el uso de RNasa A en la resuspensión final del DNA en buffer TE. El protocolo II derivó del trabajo descrito por Aljanabi et al. (1999) sin modificaciones. Los ensayos se realizaron en el laboratorio de Biotecnología de plantas del Campo Experimental Tecomán-INIFAP, Colima, México.
Material vegetal. Se recolectaron hojas sanas y jóvenes con 6 meses de edad de plantas de caña de azúcar establecidas en campo. Las muestras foliares se transportaron refrigeradas al laboratorio y se almacenaron a -70 °C hasta el momento de su uso. Para todas las muestras el DNA se extrajo a partir de 100 mg de tejido foliar. La molienda de tejido +N2 se realizó en mortero con ayuda de un pistilo; y se recuperó la cantidad requerida de tejido con una cucharilla; mientras que para la molienda -N2 se adicionó el buffer de homogeneización al tejido en el mortero y se recuperó la mezcla con micropipeta.
Protocolo I. El tejido se maceró (+N2 y -N2) y homogeneizó con 800 μL de solución amortiguadora CTAB [100 mM de Tris-HCl pH 8, 50 mM de EDTA pH 8, 1.4 M de NaCl, 3% (p/v) de CTAB y 1% (p/v) de PVP]. Posteriormente se adicionó 10 μL de 2-β-mercaptoetanol concentrado y 5 μL de proteínasa K (20 mg/mL). Se incubó las muestras durante 60 min a 65 °C en baño María y se mezcló por inversión cada 5 minutos. Se adicionó 800 μL de la solución fenol:cloroformo:alcohol isoamílico en una relación de 25:24:1, se mezcló vigorosamente usando un vórtex y se centrifugó a 14 000 rpm a 4 °C durante 10 min.
El sobrenadante recuperado se colocó en tubos de 1.5 mL y el DNA se precipitó con 0.6 volúmenes de isopropanol frío y 0.1 volúmenes de acetato de sodio 3 M pH 5.2 durante 30 min a -20 °C. La mezcla se centrifugó a 14 000 rpm a 4 °C durante 10 min. El sobrenadante se decantó y se agregó 500 μL de etanol al 70%; se agitó manualmente hasta desprender la pastilla de DNA y se centrifugaron los tubos a 14 000 rpm a 4 °C durante 5 min, se decantó la fase acuosa y se invirtieron sobre papel absorbente para retirar el exceso de etanol. La pastilla de DNA se secó a temperatura ambiente y resuspendió en 40 μL de amortiguador TE (10 mM de Tris- HCl, 1 mM de EDTA pH 8). Finalmente se agregó 1 μL de RNasaA(10 mg/mL), se mezcló con micropipeta, se incubó a temperatura ambiente por 5 minutos y se almacenó a -20 °C.
Protocolo II. Se maceró el tejido (+N2 y -N2) y mezcló con 500 μL de amortiguador de homogeneización [200 mM de Tris-HCl pH 8, 50 mM de EDTA pH 8, 2.2 M de NaCl, 2% (p/v) de CTAB, 0.06% (p/v) de sulfito de sodio]. Se adicionó 100 μL de N-lauril-sarcosina al 5% (p/v), 100 μL de PVP al 10% (p/v) y 100 μL de CTAB al 20% (p/v). Se incubó a 65 °C durante 60 min en baño María y mezcló por inversión cada 5 minutos. Las muestras se enfriaron a temperatura ambiente y se agregó un volumen de la solución fenol:cloroformo:alcohol isoamílico (25:24:1). Se mezcló por inversión y se centrifugó a 14 000 rpm durante 10 min a 4 °C. Se recuperó la fase acuosa y transfirió a tubos estériles. Se agregó un volumen de isopropanol frío seguido de 100 μL de NaCl 6 M y se incubó a -20 °C por 60 min. Posteriormente, se centrifugaron los tubos durante 10 min a 14 000 rpm. La pastilla de DNA se lavó con 1 mL de etanol al 70% y se secó a temperatura ambiente. Se resuspendió el DNA en 40 μL de amortiguador TE (10 mM de Tris-HCl, 1 mM de EDTA, pH 8) y se almacenó a -20 °C.
Cuantificación, pureza e integridad del DNA. La concentración de DNA (ng/μL) se cuantificó con un espectrofotómetro NanoDrop Thermo Scientific y se convirtió a mg/g. Como criterio de pureza se consideraron las relaciones de A260:280 y A260:230. La integridad de los ácidos nucleicos se verificó mediante la separación por electroforesis en gel de agarosa al 1% de 1 μg de DNA genómico, se tiñó con bromuro de etidio (10 mg/mL) y visualizó con luz ultravioleta en un foto documentador.
Funcionalidad del DNA purificado
Marcador RAPD. Se utilizó el oligonucleótido OPA-10- 5′-GTGATCGCAG-3’(Pan et al., 1996). El volumen de la reacción de PCR fue de 25 μL conteniendo amortiguador de reacción 1 X, 1.5 mM de MgCl2, 1 μM de cada oligonucleótido, 100 μM de dNTPs, 1.25 U de ADN polimerasa (BioTecMol) y 50 ng deADN genómico. Las condiciones de amplificación para la reacción se llevaron se basaron de acuerdo a Chen et al., 2011, con algunas modificaciones: desnaturalización inicial a 94 °C-4 min, seguido de 45 ciclos de 94 °C-1 min, 36 °C-1 min y 72 °C-2 min de desnaturalización, alineamiento y extensión, respectivamente, y una extensión final a 72 °C-7 min. Los productos de PCR se separaron mediante electroforesis horizontal en geles de agarosa al 1.5% (p/v) con amortiguador TAE teñidos con bromuro de etidio (10 mg/mL).
Marcador TRAP. Se utilizó el juego de oligonucleótidos Arbi 3 5 ́-GACTGCGTACGAATTTGA-3 ́ y SuSy -5′-GGAGGAGCTGAGTGTTTC-3’ (Hu y Vick, 2003; Devarumath et al., 2013). La reacción de PCR se llevó a cabo en un volumen de 25 μL conteniendo amortiguador de reacción 1 X, 1.5 mM de MgCl2, 1 μM de cada oligonucleótido, 100 μM de dNTPs, 1.25 U de DNA polimerasa (BioTec Mol) y 50ng de DNA genómico. Las condiciones de amplificación para la reacción fueron las siguientes: desnaturalización inicial a 94 °C-2 min, seguido de 5 ciclos de 94 °C-45 s, 35 °C-45 s y 72 °C-1 min de desnaturalización, alineamiento y extensión, respectivamente; seguido por 35 ciclos de 94 °C-45 s, 50 °C-45 s y 72 °C-1 min de desnaturalización, alineamiento y extensión, respectivamente. La extensión final fue de 72 °C-7 min (Hu y Vick, 2003). Los productos de PCR se separaron mediante electroforesis horizontal en geles de agarosa al 1.5% (p/v) con amortiguador TAE y se tiñeron con bromuro de etidio (10 mg/mL).
Análisis estadístico. El rendimiento (mg/g) y pureza (A260:280 y A260:230) del DNA se analizaron para un análisis de varianza utilizando un diseño completamente al azar con arreglo factorial 2 (protocolos) x 2 (+N2 y -N2), dando un total de 4 tratamientos y 10 repeticiones. Se consideró como unidad experimental una muestra de DNA de caña de azúcar. Se utilizó la prueba de Duncan de rangos múltiples para comparar los valores medios entre las variables al 95% de probabilidad mediante el paquete estadístico SAS. La integridad y funcionalidad del DNA fueron evaluadas visualmente con geles de agarosa.
Resultados y discusión
Rendimiento del DNA
El efecto del uso del N2 sobre el rendimiento del DNA obtenido a partir de hojas de caña de azúcar se muestra en el Cuadro 1. Los valores medios de rendimiento del DNA extraído +N2 (0.68 mg/g), fueron significativamente superiores a los obtenidos sin el uso del mismo (0.50 mg/g) (p˃ 0.05). Estos resultados superan a los reportados por otros autores, quienes extrajeron DNA de caña de azúcar, sin el uso de N2 y con rendimientos de DNA inferiores a los 0.30 mg/g de tejido foliar (Honeycutt et al., 1992; Hossain et al., 2006; Vaze et al., 2010).

a, b, c Medias con diferente superíndice entre columnas y filas denotan diferencias significativas (p< 0.05). A, B Medias con diferente superíndice en la columna denotan diferencias significativas entre protocolos (p< 0.05). M, N Medias con diferente superíndice en la fila denotan diferencias significativas con el uso del N2 (p< 0.05).
Cuadro1. Rendimiento del DNAextraído de hojas jóvenes de S. officinarum (+N2= con nitrógeno líquido; -N2= sin nitrógeno líquido).
Los resultados obtenidos en el presente estudio indican que el uso de N2 eleva el rendimiento de DNAde S. officinarum. De forma general, el empleo de N2 está ampliamente documentado en plantas (Ahmad et al., 2004; Niu et al., 2008;Arif et al.,2010; Sharma et al., 2010; Cingilli-Vural y Dageri, 2011; Doosty et al., 2012; Huaqiang et al., 2013a; Huaqiang et al., 2013b); hongos (Niu et al., 2008; Motkova y Vytrasová, 2011; Cáceres et al., 2012; Prabha et al., 2013) y animales (Gross-Bellard et al., 1973; Chen et al., 2010; Eschbach, 2012). El uso de N2 congela de -196 a -210 °C las muestras de tejido, facilitando la ruptura de todas las paredes y membranas celulares, permitiendo así la exposición de los ácidos nucleicos con el amortiguador de extracción (Cáceres et al., 2012).
Lo anterior sugiere que probablemente el uso de N2 libera una mayor cantidad de ácidos nucleicos de un mayor número de células, obteniéndose mayor rendimiento. También se ha reportado la obtención de DNA de buena calidad y elevado rendimiento en plantas -N2 (Ahmad et al., 2004; Tung-Nguyen et al., 2009; Arif et al., 2010; Sahu et al., 2012), aunque la pureza y concentración en algunos casos son más bajos (Sharma et al., 2010; Cáceres et al., 2012). En caña de azúcar la molienda de tejido -N2 es complicada, debido a la elevada cantidad de fibra que contienen las hojas. A pesar de que el N2 es caro y difícil de conseguir en algunas regiones (Aljanabi et al., 1999; Sharma et al., 2010; Sahu et al., 2012), los resultados del presente trabajo indican que es preferible utilizar N2 en la extracción de DNA de hojas de S. officinarum.
Por otra parte, se detectaron diferencias altamente significativas con el uso de los dos protocolos evaluados (p˃ 0.05) (Cuadro 1). Las diferencias entre los protocolos son: el I incluyó proteínasa K y 2-β-mercaptoetanol en el buffer de homogeneización, además utilizó acetato de sodio-isopropanol para la precipitación del DNA, así como el uso de RNasa; mientras que el II incluyó sulfito de sodio y N-lauril-sarcosina en el buffer de homogeneización y NaCl-isopropanol para la precipitación del DNA. El efecto de la proteínasa K es responsable de la lisis celular y digerir proteínas e inactivar RNasas y DNasas (Shahriar et al., 2011), mientras que el 2-β-mercaptoetanol desnaturaliza proteínas mediante el rompimiento de los puentes disulfuro. Por otra parte, el sulfito de sodio inhibe la oxidación de los polifenoles y el N-lauril-sarcosina desnaturaliza proteínas mediante el rompimiento de enlaces covalentes (Aljanabi et al., 1999). La RNasa A contenida en el protocolo I, actúa rompiendo los enlaces fosfodiéster entre las ribosas y grupos fosfato del RNA (Sambrook et al., 2001). Todos estos componentes probablemente contribuyeron a que se obtuviera un mayor rendimiento de DNA con el protocolo I. Además, el ANOVA detectó diferencias significativas en la interacción de protocolos*N2 (p˃ 0.05), por lo que el mejor tratamiento para extraer los rendimientos más elevados de DNA de hojas de caña de azúcar fue el protocolo I +N2.
Los resultados obtenidos en este estudio sobre rendimiento de DNA superan a los reportados por Honeycutt et al. (1992), quienes obtuvieron un rendimiento de DNA de 0.28 mg/g en S. officinarum utilizando un método modificado de CTAB, que incluyó el uso de espermidina, polietilenglicol (PEG) y sacarosa en el amortiguador de homogeneización. La espermidina debido a su carácter policatiónico se une con las moléculas de DNA de carga negativa, modificando su conformación y precipitándolas (Wallace et al., 2003). El PEG también precipita ácidos nucleicos, mientras que la adición de sacarosa al amortiguador aumenta el rendimiento del DNA extraído, aunque no está muy clara su interacción en el proceso de extracción de DNA (Paithankar y Prasad, 1991; Takakura y Nishio, 2012). Los resultados obtenidos sugieren que el uso de espermidina y PEG no es necesario para obtener DNAde calidad de S. officinarum, reduciendo el costo del proceso de extracción.
Porotraparte, Aljanabi et al. (1999), obtuvieron rendimientos elevados de DNA de 0.50-0.80mg/g de tejido de meristemos de caña de azúcar utilizando el equipo “Ultra-Turax” para la homogeneización de las muestras y una modificación del método del CTAB de Doyle y Doyle (1990) que incluyó altas concentraciones de CTAB (20%) y NaCl (6 M) para remover polisacáridos. En el presente trabajo se reprodujo la metodología descrita por el autor antes mencionado (protocolo II), utilizando 100 mg de tejido foliar (no meristemos). Los rendimientos de DNA obtenidos fueron muy bajos y se mantuvieron en un rango de 0.10-0.30 mg/g. Estos resultados indican que el tipo de tejido utilizado y equipo para la extracción de lDNA de S.officinarum podrían explicar las diferencias entre los rendimientos obtenidos con los dos protocolos. Por lo anterior, no se requiere utilizar altas concentraciones de CTAB y NaCl para obtener DNA con pureza elevada a partir de hojas jóvenes de S. officinarum.
Hossain et al. (2006), evaluaron tres protocolos de extracción de DNApara caña de azúcar a partir de hojas y meristemos, con una modificación en la metodología propuesta por Aljanabi et al. (1999). Los autores sustituyeron el N-lauril-sarcosina por dodecilsulfato sódico (SDS), ambos detergentes aniónicos que rompen los enlaces covalentes entre las proteínas, desnaturalizándolas en el proceso de purificación del DNA. Los autores obtuvieron un rendimiento de DNA de 0.04 mg/g, valores significativamente más bajos a los aquí reportados para hojas (0.20 mg/g) (Cuadro 1), utilizando el protocolo II. El uso de distintos detergentes aniónicos podría tener diferente efectividad sobre el rompimiento de los enlaces covalentes de las proteínas, por lo que probablemente el uso de N-lauril-sarcosina podría aumentar los rendimientos de DNA de S. officinarum con el protocolo II. Por otra parte, Vaze et al. (2010) reportaron rendimientos bajos de DNA de 0.02-0.10 mg/g en tejido foliar seco de caña de azúcar.
Estas variaciones entre rendimientos pueden ser explicadas por el tipo y condición de tejido utilizado, ya que en el presente estudio se emplearon hojas jóvenes, las cuales están en constante división celular, por lo que hay mayor cantidad de células por gramo de tejido; además, la concentración de compuestos fenólicos, polisacáridos y metabolitos secundarios es menor (Jobes et al., 1995). Honeycutt et al. (1992) usaron hojas maduras, donde la concentración de compuestos fenólicos, polisacáridos y metabolitos secundarios es mayor y el número de células por gramo de tejido es menor a comparación de las hojas jóvenes. Por otra parte, Aljanabi et al. (1999), incluyeron meristemos de caña de azúcar, los cuales tienen propiedades similares a las hojas jóvenes. Por su parte, Hossain et al. (2006) hicieron uso de meristemos y hojas; mientras que Vaze et al. (2010) extrajeron el DNA de hojas secas de caña de caña de azúcar, donde el rendimiento y calidad del DNA son bajos y puede estar degradado. Los resultados anteriores sugieren el uso de hojas jóvenes de S. officinarum para extraer y purificar DNA de elevado rendimiento y pureza aceptable.
Pureza e integridad del DNA
El efecto del uso del N2 y los protocolos de extracción de DNA sobre la pureza (A260:A280 y A260:A230) del DNA obtenido a partir de hojas de caña de azúcar se muestra en la Cuadro 2 y 3. Una muestra pura de DNA tiene una relación de A260:A280 entre 1.8-1.9, mientras que la relación A260:A230 debe ser de 1.8-2.2 (Gallagher y Desjardins, 2011). Las purezas (A260:A280) +N2 y -N2 fueron de 2.01 y 2.00, respectivamente, por lo que no se detectaron diferencias significativas con esta variable (p˃ 0.05). Por otra parte, se detectaron diferencias altamente significativas con el uso de los protocolos (p˃ 0.05) (Cuadro 2), mientras que la interacción protocolos*N2 fue significativa (p˃ 0.05). Estos resultados son consistentes con los reportados en caña de azúcar por Aljanabi et al. (1999) y Vaze et al. (2010), mientras que Honeycutt et al. (1992) y Hossain et al. (2006) reportaron purezas por debajo de 1.8, por lo que probablemente el DNA se encuentra ligeramente contaminado por proteínas y/o fenoles (Gallagher y Desjardins, 2011; Huaqiang et al., 2013a).
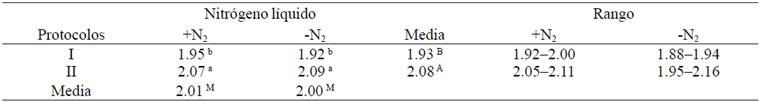
a, b, c Medias con diferente superíndice entre columnas y filas denotan diferencias significativas (p< 0.05). A, B Medias con diferente superíndice en la columna denotan diferencias significativas entre protocolos (p< 0.05). M Medias con diferente superíndice en la fila denotan diferencias significativas con el uso del N2 (p< 0.05).
Cuadro 2 Comparación de la pureza (A260:A280 nm) del DNA extraído de S. officinarum (+N2= con nitrógeno líquido; -N2= sin nitrógeno líquido).

a, b Medias con diferente superíndice entre columnas y filas denotan diferencias significativas (p< 0.05). A, B, C Medias con diferente superíndice en la columna denotan diferencias significativas entre protocolos (p< 0.05) M, N Medias con diferente superíndice en la fila denotan diferencias significativas con el uso del N2 (p< 0.05).
Cuadro 3 Comparación de la pureza (A260:A230 nm) del DNA extraído de S. officinarum (+N2= con nitrógeno líquido; -N2= sin nitrógeno líquido).
Aljanabi et al. (1999) obtuvieron para todas sus muestras A260/280 de entre 1.76-1.96 y reportaron que el RNAse degradó en el proceso de extracción, sin embargo, en este estudio con la misma metodología (protocolo II), se reportó para todas las muestras valores de la relación de A260:A280 por encima de 2 (Cuadro 2), indicativo de presencia de RNA, el cual no se degradó durante el proceso de extracción según lo descrito por Aljanabi et al. (1999), por lo que las cuantificaciones de DNA con el protocolo II podrían estar sobrestimadas. Lo anterior fue confirmado por electroforesis en gel de agarosa (Figura 1, A). Estos resultados sugieren el uso de RNasa A en el proceso de extracción de DNA. Con el uso del protocolo I la RNasa A no fue eliminada, lo anterior sugiere que puede ser posible la omisión de la incubación a 37 °C y posterior desactivación y limpieza de esta enzima, ahorrando tiempo. No se ha observado que exista inhibición por efecto de la RNasa A en las reacciones de PCR, por lo que se recomienda la inclusión de RNasa A (10 mg/mL) en la resuspensión final del DNA.

Figura 1 Electroforesis en gel de agarosa del DNA genómico de S. officinarum extraído con dos protocolos basados en el método del CTAB a partir de tejido foliar. En cada carril se cargó 1 μg de DNA. MPM= marcador de peso molecular, 1 Kb plus (Invitrogen) A) extracción +N2. Protocolo I= carriles 1-9; protocolo II= carriles 10-18. B) Extracción -N2. Protocolo I= carriles 1-4; protocolo II= carriles 5-8.
Las purezas (A260:A230) +N2 y -N2 fueron de 1.78 y 1.93, respectivamente, detectándose diferencias significativas con esta variable (p˃ 0.05), mientras que el efecto de los protocolos fue altamente significativo (p˃ 0.05) y la interacción protocolos*N2 fue significativa (Cuadro 3). Los datos reportados con el protocolo I indicaron que el DNA se encuentra libre de polisacáridos. Los métodos de extracción de DNA específicos para S. officinarum descritos por Honeycutt et al. (1992), Aljanabi et al. (1999), Hossain et al. (2006) y Vaze et al. (2010) no reportan la relación de A260:A230 como segundo criterio de pureza del DNA, a la cual se detectan los polisacáridos. El protocolo II presentó valores inferiores de A260:230 de 1.8 (Cuadro 3), por lo que probablemente los DNA se encuentran ligeramente contaminados por polisacáridos o fenoles. Se recomienda el uso del protocolo I +N2 para extraer DNA de hojas de S. officinarum ya que ambos criterios de 1-9 pureza (A260:A280 y A260:A230) resultaron óptimos.
La integridad del DNA genómico influenciada por el uso del N2 y los dos protocolos de extracción en hojas jóvenes de caña de azúcar se observa en la Figura 1. Los geles de agarosa con DNA extraído de hojas que fueron molidas con N2 mostraron patrones de bandas de DNA genómico de elevado peso molecular en todos los carriles con los dos protocolos evaluados (Figura 1-A), por lo que no se detectó degradación del DNA genómico. Las muestras del protocolo II (Carriles 10-18) mostraron contaminación por RNA debida a la omisión del tratamiento con RNasa A según lo descrito por Aljanabi et al. (1999). El protocolo I resultó ser el mejor para obtener DNA íntegro y sin residuos de RNA(Figura 1-A, carriles 1-9). El DNA genómico obtenido tiene una integridad similar a la reportada por Aljanabi et al. (1999) y Vaze et al. (2010) en caña de azúcar y a la de otros autores que utilizaron el método del CTAB en frijol caupí (Huaqiang et al., 2013a; Huaqiang et al., 2013b). Por otra parte, el DNA extraído -N mostró patrones de DNA barrido con el uso de los dos protocolos (Figura 1-B), indicativo de degradación del mismo. Resultados similares fueron obtenidos por Honeycutt et al. (1992). Se recomienda el uso de N2 para extraer DNA con integridad aceptable de hojas de S. officinarum.
Funcionalidad
La Figura 2 muestra los perfiles de dos marcadores moleculares utilizados para verificar la funcionalidad del DNA aislado con los protocolos establecidos en el presente trabajo. Para el marcador RAPD todas las muestras amplificaron perfectamente sin que se notara algún efecto por las variables N2 y protocolos (Figura 2-A-). Además, los residuos de RNA (Figura 1, A) del protocolo II no tuvieron efecto negativo en la reacción de PCR, amplificándose un buen perfil de bandas. Por otra parte, con el marcador TRAP se obtuvo un patrón de bandas de mejor calidad con las muestras que fueron tratadas +N2 (Figura 2-B, carriles 1 y 2). Las muestras extraídas -N2 presentaron un patrón de bandas difuso y de escasa calidad para su interpretación (Figura 2-B, carriles 4 y 5). Es probable mejorar el perfil de bandas con la modificación de la temperatura de alineamiento (Tm) del oligonucleótido. Por otra parte, teniendo en cuenta que en los marcadores RAPD es complicada su reproducibilidad en otros laboratorios, es importante que el DNA tenga valores óptimos de A260:280 y A260:230 para sobrellevar este problema; por lo que se recomienda el uso del protocolo I +N2 para extraer DNA de hojas de S. officinarum para su uso con marcadores moleculares.
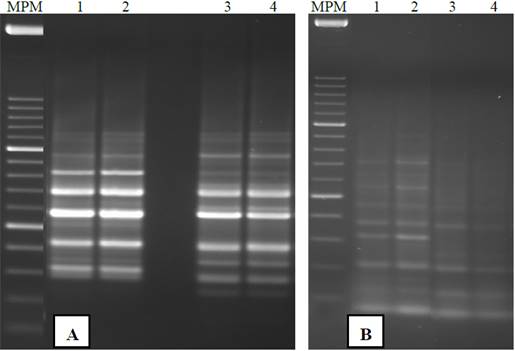
Figura 2 Marcadores moleculares con el DNA de hojas de S. officinarum extraído con dos protocolos basados en el método del CTAB. MPM= marcador de peso molecular 100 pb (Invitrogen). A) RAPD con oligonucleótido OPA 10. B) TRAP con oligonucleótidos SuSy y Arbi 3. Carriles 1 y 2= muestras molidas +N2 con los protocolos I y II, respectivamente. Carriles 4 y 5= muestras molidas -N2 con los protocolos I y II, respectivamente.
Además de los marcadores moleculares utilizados para explorar la diversidad genética de S. officinarum, el DNA obtenido con el protocolo I +N2 ha sido usado exitosamente en metodologías basadas en PCR y secuenciación para detección de fitopatógenos en caña de azúcar: Xanthomonas albilineans, Leifsonia xyli subsp. xyli, Puccinia melanocephala y P. kuehnii causantes de enfermedades de importancia económica: escaldadura, raquitismo, roya café y roya naranja, respectivamente. También se ha utilizado en PCR tiempo real para la detección de Candidatus Liberibacter asiaticus, agente causal del Huanglongbing, en varias especies de cítricos (Citrus aurantifolia, C. limon y citranges) (datos no mostrados).
Conclusiones
Las modificaciones realizadas al método del CTAB para la extracción de DNA de S. officinarum permitieron optimizar un protocolo para obtener DNA de buena calidad y elevada cantidad. Los mayores rendimientos de DNA se obtuvieron con la interacción del protocolo I +N2 para la molienda del tejido foliar. Además este mismo protocolo consumió el menor tiempo.
La pureza (A260:280 y A260:230) del DNA aislado fue de mayor calidad con el uso del protocolo I +N2. La integridad del DNA no presentó degradación +N2, mientras que la degradación se presentó en muestras de DNA extraídas -N2 con los dos protocolos evaluados.
En cuanto a la funcionalidad del DNA se obtuvieron perfiles de bandas nítidas y bien marcadas con el uso del marcador molecular RAPD +N2 y -N2. Mientras que para el marcador TRAP se logró un mejor patrón de bandas con la inclusión de N2 en cualquiera de los dos protocolos. Se recomienda emplear el protocolo I +N2 para la extracción de DNA de hojas de caña de azúcar que podría ser utilizado con diversos marcadores moleculares en programas de mejoramiento genético.

