











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
En la actualidad, la mayor parte de la energía se obtiene mediante la combustión de combustibles fósiles (Mortimer, 1991). Los combustibles fósiles se han vuelto escasos, su explotación es más costosa, no son renovables y contaminan el ambiente. Debido a esto se ha optado por el desarrollo de alternativas para la generación de energía (Demirbas, 2005). La producción de biogás es un proceso natural que ocurre en forma espontánea en un entorno anaeróbico. Dicho proceso lo realizan microorganismos como parte del ciclo biológico de la materia orgánica, el cual involucra la fermentación o digestión de materiales orgánicos para obtener el biogás (Demirel et al., 2008).
Los biodigestores son sistemas diseñados para optimizar la producción de biogás a partir de desechos agrícolas, estiércol o efluentes industriales, los cuales permiten así la obtención de energía limpia y de bajo costo a partir de una fuente renovable (Demirel et al., 2008). El rendimiento de un biodigestor está ligado principalmente a la estructura de la comunidad microbiana presente en el mismo (Demirel y Scherer, 2008). Por lo anterior, es de gran interés analizar la estructura y dinámica poblacional de estas comunidades microbianas.
El proceso bioquímico para la producción anaeróbica de metano es muy complejo. La diversidad y abundancia de microorganismos involucrados en el proceso juegan el papel más importante del proceso. A su vez, estos factores están influenciados por el tipo de materiales fermentables, las variaciones climáticas, el diseño de los reactores, entre otros. Los pasos iniciales del proceso de producción anaeróbica de metano comienzan con la reducción, mediada por bacterias hidrolíticas Firmicutes, de macromoléculas como proteínas, grasas complejas y carbohidratos, a sus intermediarios básicos, como aminoácidos, cadenas de ácidos grasos y monoazúcares. Posteriormente, otras bacterias (como acidógenas y acetógenas) continúan el proceso de reducción a otros componentes más básicos como acetato, dióxido de carbono e hidrógeno. Después, los microorganismos metanógenos (del dominio de las Arqueas) convierten los componentes producidos por las bacterias en metano a través de las rutas metabólicas aceticlástica e hidrogenotrófica (Thauer et al., 1993).
El entendimiento de la composición, la estructura y la dinámica de las comunidades microbianas presentes en reactores para la generación de biogás, es crucial para el desarrollo de nuevas estrategias de fermentación y para el diseño de bioreactores que incrementen el rendimiento de la producción de metano. Un estudio metagenómico puede enfocarse en la secuenciación total (Hess et al., 2011) o parcial (Logares et al., 2013) de todos los genomas presentes, o sólo en conocer la diversidad, en cualquiera de los niveles taxonómicos, de una comunidad microbiana.
El conocimiento de la diversidad microbiana es un buen primer ensayo en el proceso de caracterizar una estructura microbiana. Para ello existen diversas estrategias, una de las más comunes utiliza genes con una alta tasa de aparición de mutaciones, lo que permite que dos especies genéticamente relacionadas difieran en dicho gen. Los genes más comunes en el dominio de las arqueas y bacterias son el gen que codifica la subunidad 16 s de la ARN polimerasa, y para el caso de los eucariontes, los espaciadores intergénicos tales como ITS1 y ITS2. El gen 16 s, posee nueve regiones internas que presentan mayor variación nucleotídica con respecto al resto de otras regiones. Dichas regiones hipervariables varían en longitud desde las 100 pb hasta las 1000pb (Whiteley et al., 2012).
Con el desarrollo de la secuenciación masiva de ADN, ha sido posible identificar las comunidades microbianas presentes en bioreactores para la generación de metano. Aunque se conoce a detalle que los dos grupos básicos de microorganismos que se encuentran en las plantas fermentadoras son bacterias (que descomponen estructuras complejas) y arqueas (que producen metano), los estudios realizados hasta el momento señalan diferencias importantes en los órdenes, familias, géneros y especies específicos en función del material vegetal utilizado.
Schlüter et al. (2008) analizaron el metagenoma, mediante tecnología 454, de un bioreactor alimentado con ensilado de maíz, centeno y pollinaza. Una porción significativa de las secuencias obtenidas correspondió con la arquea Methanoculleus marisnigri; asimismo, se encontraron numerosas secuencias de genomas bacterianos de la clase Clostridia, los cuales se caracterizan por participar en el proceso de hidrólisis del sustrato. Por otro lado, Kröber et al. (2009) encontró una sobre representación de secuencias pertenecientes a la arquea Methanoculleus bourgensis, así como diversos órdenes del filum Firmicutes (en el que se encuentran los clostridiales).
Li et al. (2013) obtuvieron el metagenoma de una planta de generación de metano basada en gallinaza, estiércol de cerdo y diversos lodos. Similar a los estudios descritos anteriormente, Li et al. (2013)) encontraron una sobre representación de los clostridiales (Firmicutes), que como se mencionó contribuyen a la degradación de celulosas y proteínas. Otras especies importantes de bacterias para la descomposición de grasas y carbohidratos fueron Bacilli, Gammaproteobacteria y Bacteroidetes (pertenecientes a los Firmicutes, Proteobacteria, y Bacteroidetes, respectivamente). Las especies bacterianas dominantes fueron de seis géneros: Clostridium, Aminobacterium, Psychrobacter, Anaerococcus, Syntrophomonas, y Bacteroides. Dentro de las arqueas se identificaron los géneros Methanosarcina, Methanosaeta y Methanoculleus. Las especies de arqueas dominantes fueron Methanosarcina barkeri fusaro, Methanoculleus marisnigri JR1, y Methanosaeta theromphila.
En el presente trabajo se identificó, mediante metagenómica, una gran cantidad de microorganismos presentes en una muestra procedente de un biodigestor de nopal.
Materiales y métodos
Características del biodigestor y recolección de muestras
El biodigestor del cual fueron obtenidas las muestras consistió en garrafón de 10 L. Inicialmente se mezclaron en 10 L de agua corriente, 5 kg de nopal verdura y 0.5 kg de estiércol de vaca. La mezcla fue depositada en un contenedor y éste fue sellado. En el contenedor, se instaló una válvula de escape y una bolsa de hule para detectar la generación de gas. El día en que se realizó la mezcla inicial se tomó la primer muestra (T1); se tomaron 50 ml de la mezcla y se colocaron en un frasco copro estéril, posteriormente se selló y se almacenó a -80°C. Al tercer día (48 horas después), se tomó una segunda muestra (T2), al quinto día (96 horas después) se tomó una tercera (T3), y a los siete días (144 horas después) se tomó una última muestra (T4). En el séptimo día, se detectó la presencia de metano al observar una f lama en la válvula del biodigestor al acercar un cerrillo a ésta.
Extracción de ADN
La extracción de ADN de las cuatro muestras se realizó mediante dos estrategias, fenol-cloroformo, y con un kit comercial (EZ-10 SPIN column genomic DNA mini-preps kit for bacteria, marca Biobasic). La extracción con fenol- cloroformo se realizó de acuerdo a Wilson (1990) con algunas modificaciones. Brevemente, se descongeló la muestra y se colocó 1 ml en un tubo Eppendorf, se centrifugó a 12 000 rpm por 10 min y se separó el sobrenadante del precipitado. Se adicionaron 175 μl de TE 50:20 a la muestra y se agitó en un Vortex. Posteriormente, se agregaron 5 μl de proteinasa K (20 mg mL-1) y 20 μl de dodecil sulfato de sodio (SDS) al 10%. La muestra se mezcló levemente y se incubó a 56°C durante 2 h. Después, se agregaron 500 μl de TE 10:1 y 20 μl de NaCl 5M y se agitó por inversión. Se agregaron 500 μl de fenol frío (equilibrado), y se agitó continuamente por 10 minutos para homogeneizar. La muestra se centrifugó a 12 000 rpm a 4°C por 5 min y se recuperó el sobrenadante en un tubo nuevo.
Posteriormente, se agregaron 500 μl de cloroformo-alcohol isoamílico (24:1), se agitó suavemente por inversión, se centrifugó a 1 2000 rpm por 2 min a 4°C. Se recuperó el sobrenadante en un tubo nuevo y se repitió la extracción en cloroformo-alcohol isoamílico. Se agregaron 500 μl de isopropoanol frio y se incubaron las muestras a -20°C durante toda la noche. Después, se centrifugó la muestra a 1 2000 rpm por 1 minuto a 4°C y se decantó el sobrenadante. Finalmente, se lavó el precipitado con 1 ml de etanol al 70% frío, se centrifugó a 1 2000 rpm por 1 min a 4°C y se eliminó el sobrenadante. El precipitado de ADN se dejó secar a 65°C por dos horas y se resuspendió en 50 μl de agua destilada estéril. La extracción de ADN mediante el kit se realizó de acuerdo a las instrucciones del fabricante. La calidad del ADN se verificó en un gel de agarosa al 1.2% y se optó por utilizar únicamente las muestras de ADN extraídas mediante fenol-cloroformo y provenientes del sobrenadante.
Amplificación de regiones hipervariables V3 y V6
La amplificación de las regiones hipervariables V3 y V6 se realizó con ocho pares de iniciadores. Los iniciadores se sintetizaron de acuerdo a los requerimientos del equipo IonTorrent en el que se secuenciaron las muestras. Cada uno de los iniciadores consistió inicialmente en: a) un adaptador con la secuencia CCATCTCATCCCTGCGTGTCTCCGACTCAG (que se une a la nanoperla del secuenciador), una secuencia de reconocimiento para control del secuenciador con los nucleótidos GAT, un código de barras de 10 pb, y la región complementaria a la región hipervariable (20 pb para el iniciador forward y 17 para el reverse). Se utilizaron cuatro códigos de barra distintos, uno para cada tiempo muestreado, y dos pares de regiones complementarias, una para la región V3 y otra para la región V6. En Cuadro 1 se muestran los iniciadores utilizados.
Cuadro 1 Iniciadores utilizados para la amplificación de regiones hipervariables V3 y V6 del gen ribosomal 16s.

De acuerdo a Whiteley y colaboradores (2012), la reacción de PCR se realizó en un volumen de 50 μl conteniendo 200 μM de dNTPs, 0.5 μM de cada iniciador, 2 unidades de Phusion High-Fidelity DNApolimerasa y Phusion HF Buffer 1X (que incluye 1.5 mM de MgCl2). El programa de PCR utilizado para la amplificación de regiones hipervariables fue el siguiente: 94 oC por 5 min, seguido por 25 ciclos de 94°C por 40 s, 42°C por 1 min y 72°C por 2 min, con una extensión final de 72°C por 4 min. Los productos de PCR se separaron en un gel de agarosa al 1.2% (P/V), utilizando una cámara horizontal de electroforesis a 90V por 50 min. Una vez corroborado el peso molecular esperado de las regiones hipervariables, se enviaron muestras de cada uno de los tiempos y regiones evaluadas al Centro Nacional de Recursos Genéticos (CNRG) del Instituto Nacional de Investigaciones Forestales, Agrícolas y Pecuarias (INIFAP), para su secuenciación.
Secuenciación masiva en IonTorrent
El archivo de salida del secuenciador IonTorrent (con extensión.bam) se cargó en una estructura de datos de Matlab R2013a. El procesamiento de secuencias consistió en lo siguiente. Se eliminaron las secuencias que no contuvieran el código de barras. Por la estructura de los iniciadores utilizados, las secuencias obtenidas tenían inicialmente el código de barras (de 10 pares de bases) y posteriormente 20 pb correspondientes a la región complementaria de la región hipervariable (del iniciador forward). En este sentido, se seleccionaron únicamente secuencias de al menos 130 pb, para posterior al rasurado de secuencia para eliminar el código de barras y la región complementaria, se tuvieran secuencias útiles de al menos 100 pb. Después, se clasificaron las secuencias de acuerdo al código de barras, con el fin de clasificarlas por cada uno de los tiempos muestreados. Con las secuencias resultantes y separadamente para cada uno de los tiempos muestreados, se agruparon las secuencias repetidas, se ordenaron con base al número de repeticiones, y se seleccionaron las 200 secuencias más representadas. Las secuencias finales se compararon contra la base de datos SILVA (SILVA, http://www.arb-silva.de/aligner) para la obtención de la clasificación taxonómica.
Resultados y discusión
Extracción de ADN y amplificación de regiones hipervariables
La extracción de ADN de cada una de las muestras obtenidas del biodigestor de nopal se realizó mediante la estrategia fenol-cloroformo y con un kit comercial. La calidad del ADN extraído se evaluó en un gel de agarosa al 1.2%. Debido a que se detectó una banda de alto peso molecular más intensa y con poco barrido, se optó por utilizar las muestras de ADN obtenidos con fenol-cloroformo. En la Figura 1, los primeros ocho carriles (a-h) corresponden a los fragmentos amplificados a partir de ADN extraído mediante fenol-cloroformo, y los últimos (i-p) a los fragmentos correspondientes a la extracción mediante el kit comercial. Para cada método de extracción se tienen ocho carriles dado que cada tiempo se corrió para ambas regiones hipervariables; en este sentido, los carriles a y b corresponden al tiempo T1 de las regiones V3 y V6, respectivamente; c y d corresponden a T2 de V3 y V6, respectivamente; e y f al T3, y g y h al T4, todos para el ADN extraído mediante la metodología de fenol- cloroformo. Este arreglo se mantuvo en el resto de los carriles (i-p), correspondiente al ADN extraído mediante el kit comercial.

Secuenciación de regiones hipervariables V3 y V6
La concentración de los productos de PCR amplificadores de las regiones V3 y V6 fue ajustada a 20 picoMoles⋅μl-1 y fueron enviados al CNRG para su secuenciación. La secuenciación se realizó en un equipo IonTorrent, de Life Technologies. Cada uno de los cuatro tiempos evaluados se combinó y se obtuvieron dos muestras finales para las regiones V3 y V6. El llenado de nanoperlas de cada uno de los chips de IonTorrent se muestra en la Figura 2.
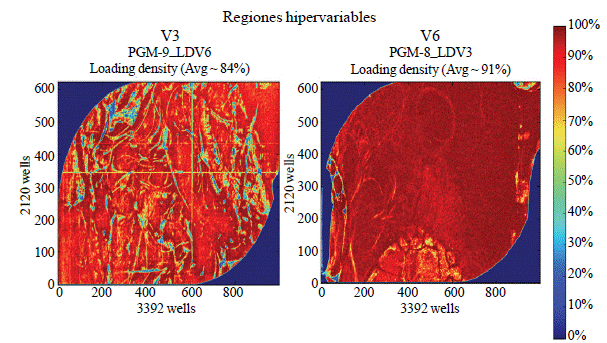
Para la región hipervariable V3 se obtuvieron 889 777 secuencias, y para la región V6 145 080. Las secuencias de 600 mayor longitud fueron de 293 y 281 pb para las regiones V3 y V6 respectivamente. La mediana del tamaño de secuencia para V3 fue de 180 pb y 135 pb para la región V6.
Clasificación de secuencias y filtrado
Como se mencionó en la sección de materiales y métodos, se utilizó un código de barras distinto para cada uno de los tiempos muestreados. Dado que la secuenciación de muestras se realizó con una mezcla de todas las muestras obtenidas, el código de barras se utilizó para clasificar las secuencias. Para esto, se escribió un código en Matlab 2014b para clasificar las secuencias según su código de barras.
En el Cuadro 2, se muestra el número de secuencias finales utilizado para posteriores análisis obtenidos después de la clasificación y ordenamiento de secuencias.
Cuadro 2 Número de secuencias obtenidas (después del proceso de filtrado) para cada uno de los tiempos muestreados.

El agrupamiento de secuencias similares redujo el total de secuencias a analizar como sigue: para la región hipervariable V3, en T1, una secuencia única con una repetición, en T2 25 207 secuencias únicas, en T3 37, 833 secuencias únicas y en T4, 9,150 secuencias únicas. Las secuencias más representadas tuvieron 717, 3 647 y 4 167 secuencias para T2, T3 y T4 respectivamente. Para la región hipervariable V6, en T1, 8 secuencias únicas, en T2 199 secuencias únicas, en T3 283 secuencias únicas y en T4 64 secuencias únicas. Las secuencias más representadas tuvieron 1, 3, 7 y 10 secuencias para T1, T2, T3 y T4 respectivamente.
Comparación de secuencias contra base de datos SILVA y análisis de biodiversidad
La comparación con la base de datos de genes ribosomales 16s SILVA se realizó con las 200 secuencias únicas más repetidas para cada uno de los tiempos muestreados. Se utilizó la paquetería SILVA Incremental Aligner (SINA), en la cual se cargaron las 200 secuencias para cada uno de los tiempos muestreados. En los Cuadros 3 y 4, se muestran los grupos taxonómicos a los cuales se asignaron cada una de las 200 secuencias evaluadas. Únicamente se presentan los niveles taxonómicos de familia y género. No fue posible obtener la especie a la que pertenece cada una de las secuencias.
Cuadro 3 Identificación de familias y géneros de microorganismos basada en la comparación de las secuencias obtenidas de la región hipervariable V3 y la base de datos SILVA.

Cuadro 4 Identificación de familias y géneros de microorganismos basada en la comparación de las secuencias obtenidas de la región hipervariable V6 y la base de datos SILVA.

Del total de géneros mostrados en los Cuadros 3 y 4, algunos están presentes sólo en alguno de los tiempos muestreados, o solo en alguna de las regiones hipervariables. En el Cuadro 5, se muestra una lista general y resumida de los distintos géneros encontrados en este ensayo metagenómico, agrupados sólo por región hipervariable y en conjunto.

De acuerdo al Cuadro 5, la utilización de la región hipervariable V3 permitió la identificación de 24 géneros bacterianos, la utilización de la región hipervariable V6 permitió identificar 28 géneros bacterianos; en conjunto, en este ensayo se lograron identificar 31 distintos géneros del dominio procarionte.
Como se mencionó anteriormente, el proceso de metanogénesis está compuesto por dos principales etapas, en la primera, se degradan los compuestos orgánicos y se produce el CO2 necesario para el desarrollo de la segunda etapa de generación de metano. Se ha reportado que la primera etapa de la metanogénesis es mediada por bacterias, especialmente del filum Firmicutes (Li et al., 2013). En el presente trabajo se identificaron 205 secuencias de la región V3 y 81 secuencias de la región V6 pertenecientes al grupo de los Firmicutes.
De acuerdo con los resultados obtenidos, no fue posible encontrar ningún género del dominio de las arqueas, todos las secuencias obtenidas fueron asignadas a géneros bacterianos diversos. Aunque se ha reportado el uso de los iniciadores aquí probados para la amplificación de regiones hipervariables en arqueas, el hecho de que no se hayan asignado secuencias al dominio de las arqueas se explicaría por los parámetros de PCR empleados ya que estos pudieron no ser los más eficientes para la amplificación de las regiones V3 y V6 de arqueas. Otra posible razón pudiese ser que el muestreo se realizó cuando las condiciones internas del biodigestor eran dominantemente aeróbicas.
Conclusiones
En México, la producción de nopal ha sido destinada únicamente para el consumo humano. El cultivo de nopal requiere insumos mínimos y rinde grandes cantidades de material vegetal. Estas ventajas pueden ser muy apreciadas si se lograse diversificar el uso actual del nopal. En el presente trabajo se caracterizó el metagenoma presente en una planta experimental de generación de metano a partir de nopal. Las secuencias hipervariables V3 y V6 permitieron identificar las secuencias obtenidas hasta un nivel taxonómico de género. Los iniciadores utilizado permitieron identificar únicamente microorganismos del dominio de las procariontes; no se identificaron arqueas. De las secuencias utilizadas para el análisis final, 205 secuencias de ADN fueron asignadas al filum Firmicutes, un importante grupo de bacterias cuya función es la degradación de materia verde para la producción de dióxido de carbono. Los resultados obtenidos sugieren que es importante caracterizar la comunidad microbiana presente en las plantas para la generación de metano, así como establecer la dinámica poblacional a través del tiempo. Dicho conocimiento será importante para el diseño racional de mejores estrategias de generación de fuentes alternativas de energía.

