










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista mexicana de ciencias agrícolas
Print version ISSN 2007-0934
Rev. Mex. Cienc. Agríc vol.7 n.3 Texcoco Apr./May. 2016
Articles
Sequencing of V3 and V6 hypervariable regions of microorganisms in a digester of nopal
1 Campo Experimental Pabellón-INIFAP. Carretera Aguascalientes-Zacatecas, km. 32.5. 20660. Pabellón de Arteaga, Aguascalientes.
2 University of Wisconsin-Madison- Department of Horticulture. 1575 Linden Dr. 53706. Madison, Wisconsin, USA.
The development of clean, efficient and low cost power generation strategies is one of the most important current challenges for society. The methanogenesis, a set of metabolic processes carried out by archaea in the final stage, is used in biodigesters to generate methane gas. Control of microorganisms in a digester is decisive in the efficiency of methane production; it also lays the foundation for change, by metabolic engineering, methanogenic microorganisms and improve efficiency. In this paper we identified the microorganisms present in a sample from a digester of nopal. To identify as many microorganisms as possible a metagenomic analysis was performed to determine two hypervariable sequences of ribosomal genes 16 s. The primers used allowed only identify organisms domain of prokaryotes; archaea they not identified. The group of organisms was the most represented phylum Firmicutes, an important group of bacteria whose function is the degradation of green material for the production of carbon dioxide.
Keywords: gen 16 s; metagenomics; methane
El desarrollo de estrategias limpias, eficientes y de bajo costo para la generación de energía, es uno de los retos actuales más importantes para la sociedad. La metanogénesis, un conjunto de procesos metabólicos llevado a cabo por arqueas en la etapa final, es aprovechada en biodigestores para la generación de gas metano. El control de los microorganismos presentes en un biodigestor es determinante en la eficiencia de producción de metano; así mismo, sienta las bases para modificar, mediante ingeniería metabólica, los microorganismos metanogénicos y mejorar su eficiencia. En el presente trabajo se identificaron los microorganismos presentes en una muestra procedente de un biodigestor de nopal. Para identificar la mayor cantidad de microorganismos posible, se realizó un análisis metagenómico para determinar dos secuencias hipervariables de genes ribosomales 16 s. Los iniciadores utilizados permitieron identificar únicamente microorganismos del dominio de las procariontes; no se identificaron arqueas. El grupo de organismos con mayor representación fue el filum Firmicutes, un importante grupo de bacterias cuya función es la degradación de materia verde para la producción de dióxido de carbono.
Palabras clave: gen 16 s; metagenómica; metano
Introduction
Currently, most of the energy is obtained by combustion of fossil fuels (Mortimer, 1991). Fossil fuels have become scarce, their exploitation is more expensive, and they are not renewable and pollute the environment. Because this has been chosen for the development of alternative energy generation (Demirbas, 2005). The Biogas production is a natural process that occurs spontaneously in an anaerobic environment. This process is carried out by microorganisms as part of the biological cycle of organic matter, which involves fermentation or digestion of organic materials for biogas (Demirel et al., 2008).
The Biodigesters are systems designed to optimize the production of biogas from agricultural waste, manure or industrial effluents, which thus allow the production of clean energy and low-cost from a renewable source (Demirel et al., 2008). The performance of a digester is mainly linked to the structure of the microbial community present in it (Demirel and Scherer, 2008). Therefore, it is of great interest to analyze the structure and population dynamics of these microbial communities.
The biochemical process for anaerobic methane production is very complex. The diversity and abundance of microorganisms involved in the process play the most important role in the process. In turn, these factors are influenced by the type of fermentable materials, climatic variations, and the reactor design, among others. The initial steps of the process of anaerobic methane production starts with the reduction, by hydrolytic bacteria Firmicutes mediated, macromolecules such as proteins, complex carbohydrates and fats, their basic intermediates such as amino acids, fatty chains monosugars acids. Subsequently, other bacteria (as acidogenas and acetogenas) continue the reduction process to more basic components as acetate, carbon dioxide and hydrogen. Then methanogens microorganisms (domain Archaea) convert components produced by bacteria in methane through metabolic pathways acetoclastic and hidrogenotrofica (Thauer et al., 1993).
The understanding of the composition, structure and dynamics of microbial communities present in reactors for the generation of biogas, is crucial for the development of new strategies fermentation and for the design of bioreactors to increase the yield of methane production. A metagenomic study may focus on the full sequencing (Hess et al., 2011) or partial (Logares et al., 2013) of all genomes present, or only know the diversity in any taxonomic levels, a community microbial.
Knowledge of microbial diversity is a good first test in the process of characterizing a microbial structure. For this there are several strategies, one of the most common uses genes with a high rate of appearance of mutations, which allows two genetically related species differ in that gene. The most common genes in the domain of archaea and bacteria are the gene encoding subunit 16 s RNA polymerase, and in the case of eukaryotes, the intergenic spacers ITS1 and ITS2 like. The gene 16 s, has nine internal regions with higher nucleotide variation relative to the rest from other regions. Said hypervariable regions vary in length from 100 bp to the 1 000 pb (Whiteley et al., 2012).
With the development of DNA sequencing mass has been possible to identify microbial communities present in bioreactors for methane generation. Although it is known in detail the two basic groups of microorganisms found in the fermenting plants are bacteria (which break down complex structures) and archaea (producing methane), studies so far indicate significant differences in orders, families, genera and species specific depending on the plant material used.
Schlüter et al. (2008) analyzed the metagenome, by 454 technology, a bioreactor fed corn silage, rye and chicken manure. A significant portion of the sequences obtained corresponded with bows Methanoculleus marisnigri; also numerous bacterial genomes sequences of the Clostridia class, which are characterized by participating in the process of substrate hydrolysis was found. Furthermore, Kröber et al. (2009) found overrepresented sequences belonging to the bows Methanoculleus bourgensis and Firmicutes phylum various commands (which are clostridial).
Li et al. (2013) obtained the metagenome of methane generation plant based on manure, pig manure and various sludge. Similar to the studies described above, Li et al. (2013) found the clostridial overrepresented (Firmicutes), which as mentioned contribute to the degradation of cellulose and proteins. Other important species of bacteria for the decomposition of fats and carbohydrates were Bacilli, Gammaproteobacteria and Bacteroidetes (belonging to the Firmicutes, Proteobacteria and Bacteroidetes, respectively). The dominant bacterial species were six genera: Clostridium, Aminobacterium, Psychrobacter, Anaerococcus, Syntrophomonas, and Bacteroides. Within archaea genera Methanosarcina, Methanosaeta and Methanoculleus were identified. The dominantarchaeawere Methanosarcina barkeri fusaro, Methanoculleus marisnigri JR1, and Methanosaeta theromphila.
In this paper identified by metagenomics, a large number of microorganisms present in a sample from a digester of nopal.
Materials and methods
Features digester and sample collection
The digester which were obtained samples consisted of 10 L. Initially carboy were mixed in 10 L of tap water, 5 kg of nopal and 0.5 kg of cow dung. The mixture was placed in a container and this was sealed. In the container, an exhaust valve and rubber bag was installed to detect gas generation. The day the initial mixture was made the first sample (T1) was taken; 50 ml of the mixture were taken and placed in a sterile bottle copro subsequently sealed and stored at -80°C. On the third day (48 hours), a second sample (T2), the fifth day (96 hours) a third (T3) was taken, and seven days (144 hours) in a final sample was taken was taken (T4). On the seventh day, the presence of methane by observing a flame valve in the digester by bringing a hillock to it was detected.
DNA extraction
The DNA extraction of the four samples was performed by two strategies, phenol-chloroform, and with a commercial kit (EZ-10 genomic DNA SPIN column mini-preps kit for bacteria, Biobasic mark). Extraction with phenol- chloroform was performed according to Wilson (1990) with some modifications. Briefly, sample was thawed and 1 ml was placed in an Eppendorf tube, centrifuged at 12 000 rpm for 10 min and the supernatant precipitate separated. 175 μl de TE 50:20 were added to the sample and vortex. Subsequently, 5 μl of proteinase K (20 mg mL-1) and 20 μl of sodium dodecyl sulfate (SDS) at 10% were added. The sample was mixed slightly and incubated at 56 °C for 2 h. Then 500 μl TE were added 10: 1 and 20 μl NaCl 5M and stirred by inversion. A 500 μl of cold phenol (balanced) were added and continuously stirred for 10 minutes to homogenize. The sample was centrifuged at 12 000 rpm at 4 °C for 5 min and the supernatant was recovered in a new tube.
Subsequently, 500 μl of chloroform-isoamyl alcohol (24:1) were added, stirred gently by inversion, centrifuged at 12 000 rpm for 2 min at 4 °C. The supernatant was recovered in a new tube and the extraction was repeated chloroform-isoamyl alcohol. 500 μl of cold isopropoanol were added and samples were incubated at -20 °C overnight. The sample was centrifuged 12 000 rpm for 1 minute at 4 °C and the supernatant decanted. Finally the precipitate with 1 ml of cold 70% ethanol washed, centrifuge dat2000rpm for 11min at 4°C and the supernatant was removed. The DNA precipitate was allowed to dry at 65 °C for two hours and resuspended in 50 μl of sterile distilled water. The DNA extraction was performed using the kit according to the manufacturer's instructions. DNA quality was verified in an agarose gel 1.2% and chose to use only the DNA samples extracted by phenol-chloroform and from the supernatant.
Amplification hypervariable regions V3 and V6
The amplification of V3 and V6 hypervariable regions was performed with eight pairs of primers. The primers were synthesized according to the requirements of IonTorrent computer that samples sequenced. Each of the initiators initially consisted of: a) an adapter with CCATCTCATCCCTGCGTGTCTCCGACTCAG sequence (which binds to nanoperla sequencer), a recognition sequence for control sequencer with GAT nucleotides, a bar code 10 bp, and the region complementary to the hypervariable region (20 bp for the forward primer and the reverse 17 for). Four different bar codes, one for each sampled time, and two pairs of complementary regions, one for the V3 region and one for the V6 region were used. Table 1 shows the primers used.

According to Whiteley et al. (2012), the PCR reaction was performed in a volume of 50 μl containing 200 μM dNTPs, 0.5 μM of each primer, 2 units of Phusion High-Fidelity DNA Polymerase and Phusion HF Buffer 1X (including 1.5 μM MgCl2). The PCR program used for amplification of hypervariable regions was as follows: 94°C for 5 min, followed by 25 cycles of 94°C for 40 s, 42°C for 1 min and 72°C for 2 min, with a final extension of 72°C for 4 min. The PCR products were separated on agarose gel 1.2% (P/V), using a horizontal electrophoresis chamber at 90V for 50 min.
Once confirmed the expected molecular weight of the hypervariable regions, samples were sent of each of the times and regions assessed the National Center for Genetic Resources (CE the National Institute of Forestry, Agriculture and Livestock (INIFAP), for sequencing.
Massive sequencing in IonTorrent
The output file IonTorrent sequencer (with extensión. bam) was loaded into a data structure of Matlab R2013a. The processing sequence was as follows. Sequences not containing the barcode is eliminated. By the structure of the primers used, the sequences obtained initially had the bar code (10 bp) and 20 bp corresponding to subsequently the complementary region of the hypervariable region (primer forward). Here, only sequences of at least 130 bp, for post- shaving selected sequence to remove the bar code and the complementary region, useful sequences of at least 100 bp were taken. Then the sequences according to the barcode were classified, in order to classify each of the sampled time. With the resulting sequences and separately for each of the sampled times, repeated sequences were pooled, they ordered based on the number of repetitions, and most represented 200 sequences were selected. End sequences were compared against the data base SILVA (SILVA, http://www.arb-silva.de/aligner) for obtaining taxonomic classification.
Results and discussion
DNA extraction and amplification hypervariable regions
The DNA extraction of each of the samples obtained from the digester of nopal was performed by phenol- chloroform strategy and with a commercial kit. Extracted DNA quality was evaluated in an agarose gel 1.2%. Because a higher intense band was detected molecular weight and little sweep, he chose to use DNA samples obtained phenol-chloroform. In Figure 1, the first eight lanes (a-h) corresponding to the amplified fragments from DNA extracted using phenol-chloroform, and the last (i-p) to the fragments corresponding to extraction with the commercial kit. For each method of extraction have eight lanes as each time ran for two hypervariable regions; in this regard, lanes a and b correspond to the time T1 regions V3 and V6, respectively; c and d correspond to T2 of V3 and V6, respectively; e and f to T3, and g and h to, all for DNA extracted by phenol-chloroform methodology. This arrangement was maintained in the rest of the tracks (i-p), corresponding to DNA extracted using the commercial kit.

Sequencing of V3 and V6 hypervariable regions
The concentration of PCR products amplifiers of V3 and V6 regions was adjusted to 20 picoMoles⋅μl-1 and were sent to the National Center for Genetic Resources of the INIFAP for sequencing. Sequencing was performed on a computer IonTorrent, from Life Technologies. Each of the four times evaluated combined and two final samples for V3 and V6 regions were obtained. The Nanobeads filling each IonTorrent chips shown in Figure 2.
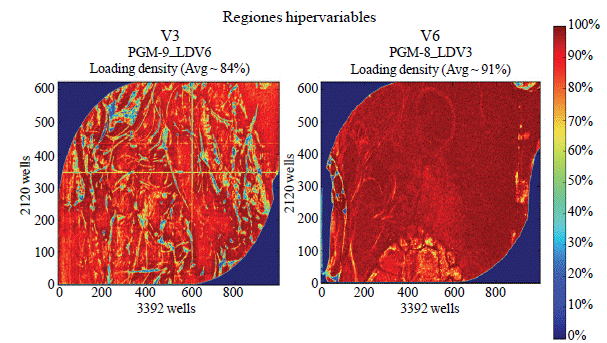
For the hypervariable region V3 sequences 889 777 were obtained, and the region V6, 145 080. The longest sequences were of 293 and 281 bp for the V3 and V6 regions respectively. The median size for V3 sequence was 180 bp and 135 bp for the V6 region.
Sequence classification and filtering
As mentioned in the section of materials and methods, a different bar code for each of the sampled times was used. Since sequencing samples was performed with a mixture of all the samples obtained, the bar code was used to classify sequences. For this, a code in Matlab 2014b was written to classify sequences by bar code.
The number of end sequences used for further analysis obtained after classification and sorting sequences shown in Table 2.
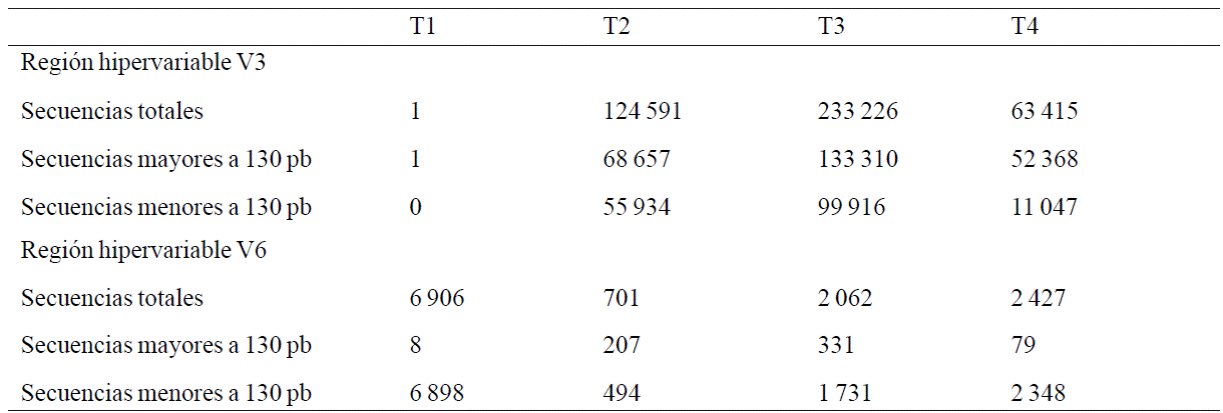
Pooling of similar sequences reduced the total sequence analyzed as follows: for the hypervariable region V3, at T1, a unique sequence with a repetition T2, 25,207 unique sequences, T3, 37, 833 unique sequences and T4, 9.150 unique sequences. The sequences most represented were 717, 3647 and 4167 sequences for T2, T3 and T4 respectively. For the hypervariable region V6, in T1, eight, unique sequences in T2, 199 unique sequences, in T3 unique sequences 283 and T4 64 unique sequences. The most represented sequences were 1, 3, 7 and 10 sequences for T1, T2, T3 and T4 respectively.
Sequence comparison against SILVA based data and analysis of biodiversity
Comparison with the database SILVA 16s ribosomal gene was performed with the 200 unique sequences more repeated for each sampled time. Incremental SILVA parcel Aligner (SINA), in which 200 sequences for each of the sampled times used were loaded. Tables 3 and 4, the taxonomic group to which assigned each of the 200 sequences shown evaluated. Only taxonomic family and genus levels are presented. It was not possible to obtain the species to which each sequence belongs.
Table 3 Identification of families and genera based on comparison of the obtained sequences of the hypervariable region V3 microorganisms and SILVA data base.

Table 4 Identification of families and genera based on comparison of the obtained sequences of the hypervariable region V6 microorganisms and SILVA data base.
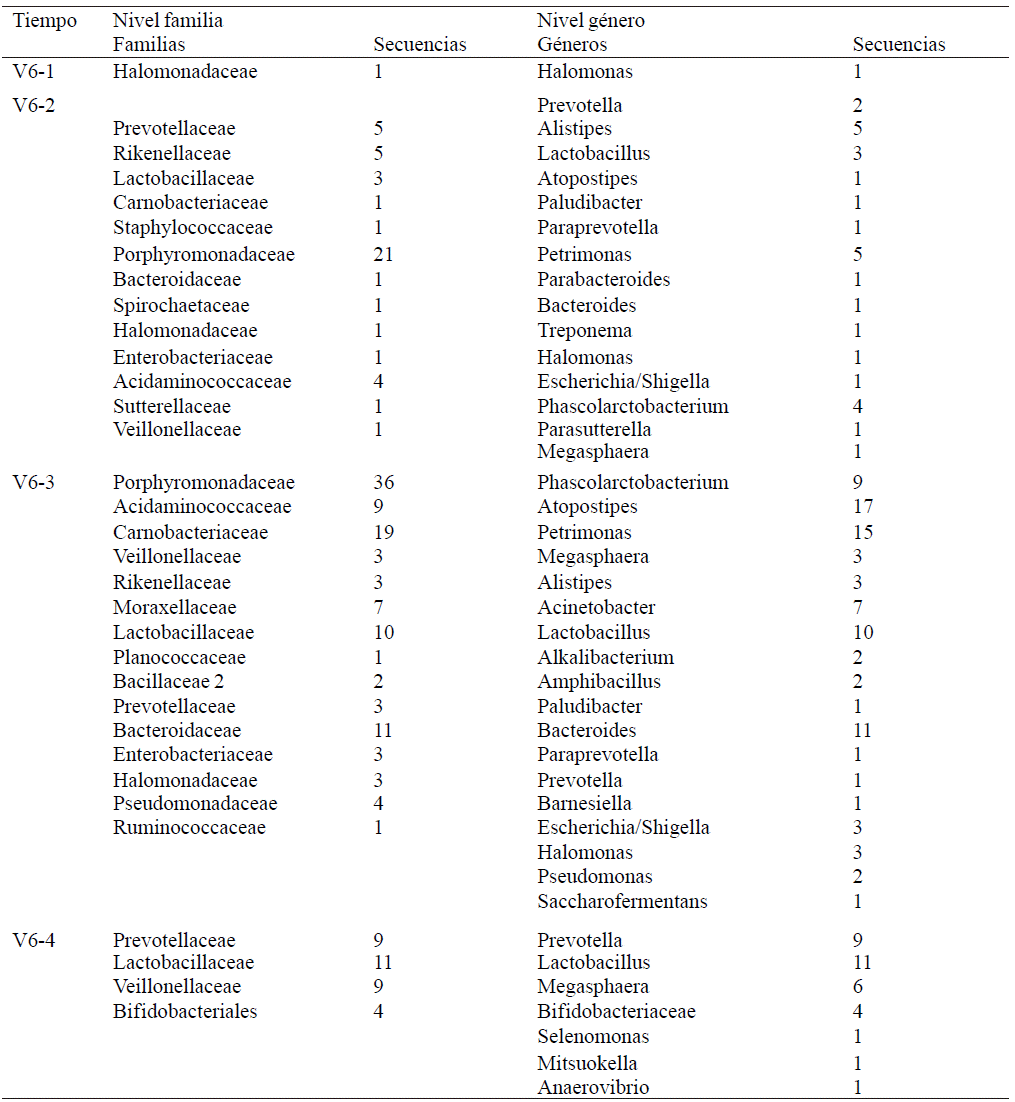
Of all genres shown in Tables 3 and 4, some are present only in some of the sampled time, or only one of the hypervariable regions. A general summary and list of different genres found in this metagenomic test only hypervariable region grouped together and shown in Table 5.
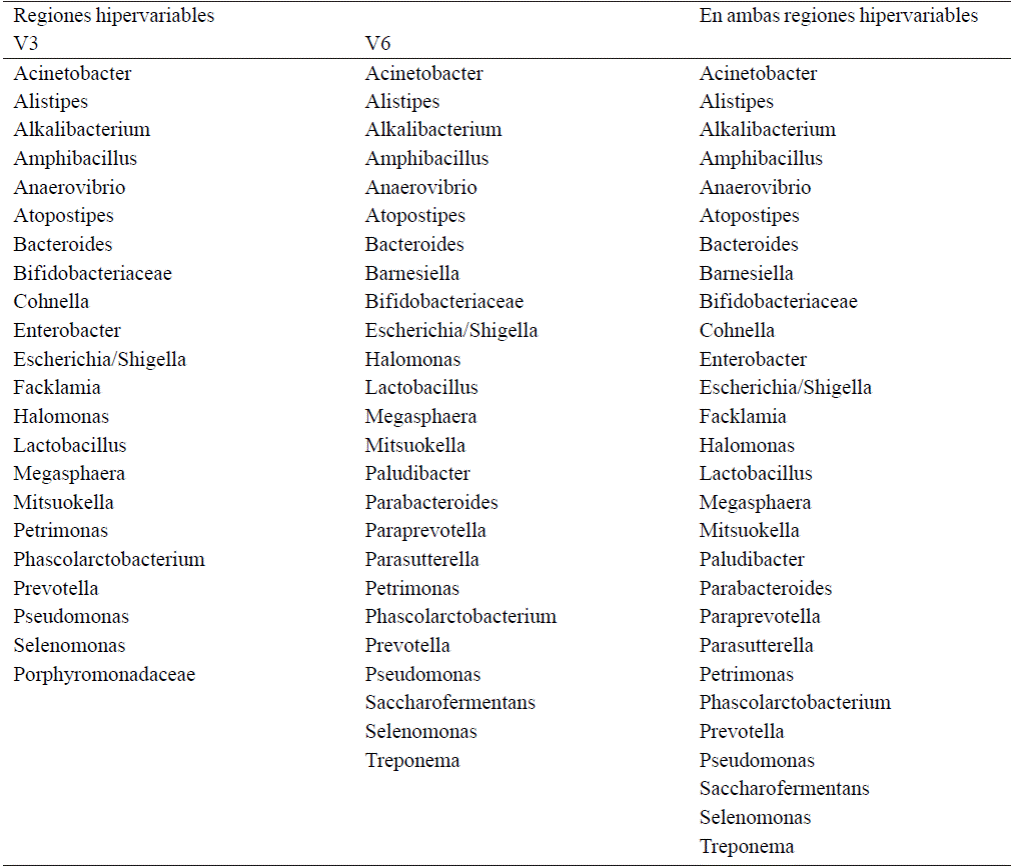
According to Table 5, the use of the hypervariable region V3 allowed the identification of 24 bacterial gender, using hypervariable region identified 28, V6 bacterial genera; together, in this trial they were able to identify 31 different genres of prokaryote domain.
As mentioned above, the methanogenesis process comprises two main stages, the first, degrade organic compounds and CO2 necessary for development of the second generation stage methane. It has been reported that the first stage of methanogenesis is mediated by bacteria, especially of the phylum Firmicutes (Li et al., 2013). In this work 205 were identified of V3 region and sequences 81 of V6 region sequences belonging to the group of the Firmicutes.
According to the results, it was not possible to find any kind of domain archaea, all the sequences obtained were assigned to various bacterial genera. Although it has been reported using the here tested for amplification of hypervariable regions in archaea initiators that have been allocated sequences to the domain of archaea be explained by PCR parameters used since these might not be the most efficient for amplification of V3 and V6 regions archaea. Another possible reason could be that the sampling was performed when internal conditions were dominantly aerobic digester.
Conclusions
In Mexico, nopal production was only intended for human consumption. The nopal cultivation requires minimal inputs and yields large quantities of plant material. These advantages can be greatly appreciated if lograse diversify the current use of nopal. In the present work this metagenome was characterized in an experimental plant for methane generation from nopal. The V3 and V6 hypervariable sequences allowed to identify the sequences obtained to a taxonomic genus level. The primers used allowed only identify organisms domain of prokaryotes; archaea they not identified. Of the sequences used for the final analysis, 205 sequences DNA were assigned to the phylum Firmicutes, an important group of bacteria whose function is the degradation of green material for the production of carbon dioxide. The results suggest that it is important to characterize the microbial community present in plants for the generation of methane and establish the population dynamics over time. This knowledge will be important for the rational design of better strategies for generating alternative energy sources.
Literatura citada
Demirbas, A. 2005. Potential application of renewable energy sources, biomass combustion problems in boiler power systems and combustion related environmental issues. Progress in Energy and Combustion Science. 31(2):171-192. [ Links ]
Demirel, B.; Neumann, L. and Scherer, P. 2008. Microbial community dynamics of a continuous mesophilic anaerobic biogas digester fed with sugar beet silage. Eng. Life Sci. 8(4):390-398. [ Links ]
Demirel, B. and Scherer, P. 2008. The roles of acetotrophic and hydrogenotrophic methanogens during anaerobic conversion of biomass to methane: a review. Rev. Environ. Sci. Biotechnol. 7(2):173-190. [ Links ]
Hess, M.; Sczyrba, Al.; Egan, R.; Tae-Wan, Kim.; Chochawala, H.; Schroth, G.; Luo, S.; Douglas, S. C.; Chen, F.; Zhang, T.; Mackie, R.I.; Pennacchio, L.A.; Tringe, S.G.; Visel, A.; Woyke, T.; Wang, Z. and Rubin, E. M. 2011. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 331(6016):463-467. [ Links ]
Kröber, M.; Bekel, T.; Diaz, N. N.; Goesmann, A.; Jaenicke, S.; Krause, L.; Miller, D.; Runte, K. J.; Viehover, P.; Puhler, A. and Schluter, A. 2009. Phylogenetic characterization of a biogas plant microbial community integrating clone library 16S-rDNA sequences and metagenome sequence data obtained by 454-pyrosequencing. J. Biotechnol. 142(1):38-49. [ Links ]
Li, A.; Chu, Y.; Wang, X.; Ren, L.; Yu, J.; Liu, X.; Yan, J.; Zhang, L.; Wu, S. and Li, S. 2013. A pyrosequencing-based metagenomic study of methane-producing microbial community in solid-state biogas reactor. Biotechnol. Bio. 17 p. [ Links ]
Logares, R.; Sunagawa, S.; Salazar, G.; Cornejo-Castillo, F. M.; Ferrera, I.; Sarmento, H.; Hingamp, P. and Ogata, H. 2013. Metagenomic 16S rDNA illumina tags are a powerful alternative to amplicon sequencing to explore diversity and structure of microbial communities. Environ. Microbiol. 16(9):2659-2671. [ Links ]
Mortimer, N. D. 1991. Energy analysis of renewable energy sources. Energy Policy. 19(4):374-385. [ Links ]
Schlüter, A.; Bekel, T.; Diaz, N. N.; Dondrup, M.; Eichenlaub, R.; Gartemann, K. H.; Krahn, I.; Krause, L.; Krömeke, H.; Kruse, O.; Mussgnug, J. H.; Neuweger, H.; Niehaus, K.; Pühler, A.; Runte, K. J.; Szczepanowski, R.; Tauch, A.; Tilker, A.; Viehöver, P. and Goesmann, A. 2008. The metagenome of a biogas-producing microbial community of a production-scale biogas plant fermenter analysed by the 454-pyrosequencing technology. J. Biotechnol. 136(1-2):77-90. [ Links ]
Thauer, R. K.; Hedderich, R. and Fischer, R. 1993. Reactions and enzymes involved in methanogenesis from CO2 and H2. Methanogensis. Ferry, J. (eds). Springer US. Boston, MA, USA. 209-252 pp. [ Links ]
Whiteley, A. S.; Jenkins, S. and Waite L. 2012. Microbial 16S rRNA ion tag and community metagenome sequencing using ion torrent (PGM) platform. J. Microbial Methods. 91:80-88. [ Links ]
Wilson, K. 1990. Preparation of genomic DNA from bacteria. In: current protocols in molecular biology. Ausubel, F.M.; Brent, R.; Kingston, R. E.; Moore, D. D.; Seidman, J. G.; Smith, J. A. and Struhl, K. (Eds.). Assoc. and Wiley Interscience. NY, USA. 241-245 pp. [ Links ]
Received: January 2016; Accepted: April 2016
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
