










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista mexicana de ciencias agrícolas
versión impresa ISSN 2007-0934
Rev. Mex. Cienc. Agríc vol.4 no.1 Texcoco ene./feb. 2013
Artículos
Tamaño de muestra para estimar expresión genética de plantas transgénicas usando pruebas de grupo*
Sample size for estimating gene expression of transgenic plants using group tests
Osval Antonio Montesinos-López1§, Abelardo Montesinos-López2, Ignacio Luna-Espinoza3 y Jesús Erasmo Montesinos-López2
1 Facultad de Telemática, Universidad de Colima. Bernal Díaz del Castillo Núm. 340, Villas San Sebastián, 28045. Colima, México. §Autor para correspondencia: oamontes2@hotmail.com.
2 Departamento de Estadística, Centro de Investigación en Matemáticas (CIMAT), Guanajuato, México. (aml_uach2004@yahoo.com.mx; shumyto@hotmail.com).
3 Universidad del Istmo-Campus Ixtepec. Cd. Ixtepec, 70110, Oaxaca, México. (iluna@bianni.unistmo.edu.mx).
*Recibido: julio de 2012
Aceptado: enero de 2013
Resumen
En las regiones sur-este y centro-oeste de México se ha detectado la presencia de maíz transgénico (GM), aun cuando los efectos de la polinización cruzada entre este maíz y variedades criollas o silvestres, como Tripsacum y teocintle, son desconocidos. Por esta razón es necesario detectar la presencia de plantas transgénicas y estimar la expresión genética media de los transgenes en los cultivos normales. Sin embargo, hacer un análisis de cada una de las plantas consumiría mucho tiempo y dinero. Una alternativa para reducir costos es utilizar la prueba de grupos. Esta técnica analiza grupos que contienen tejidos de varias plantas sin inspeccionarlas individualmente, manteniendo niveles aceptables de exactitud pero a bajo costo. Cuando la prueba de grupos se utiliza para estimar expresión genética media, es importante determinar el tamaño de muestra, número de grupos, para realizar el proceso de estimación. En este contexto y bajo condiciones de normalidad, este trabajo presenta dos procedimientos, computacional y analítico, para estimar la expresión genética media de maíz GM y se proporcionan ejemplos para mostrar la aplicación de los métodos propuestos. Adicionalmente, mediante simulación se reprodujeron diversas circunstancias que un investigador puede encontrar y se proporciona un algoritmo computacional en el programa estadístico R (R Development Core Team 2007) para crear otros escenarios. Ambos procedimientos garantizan que la amplitud del intervalo de confianza W sea menor o igual que la amplitud deseada ω con probabilidad γ. Esto se logra porque los dos métodos consideran la aleatoriedad de la varianza muestral. Sin embargo, se recomienda el uso de la fórmula propuesta, procedimiento analítico, porque es preciso y sencillo de usar.
Palabras clave: intervalo de confianza, expresión genética media, normalidad.
Abstract
In the south-east and west-center of Mexico, the presence of transgenic maize has been detected (GM), even though the effects of cross-pollination between this maize and landraces such as Tripsacum and teosinte are unknown so far. It is therefore necessary to detect the presence of transgenic plants and to estimate the average gene expression of transgenes in regular crops. However, an analysis of each and every one of the plants would consume a lot of time and money as well. An alternative to reduce costs is to use test groups. This technique analyzes groups that possess tissues of several plants without individually inspecting them, keeping acceptable levels of accuracy at a lower cost. When a test group is used to estimate the average genetic expression it is important to determine the sample size and the number of groups for the estimation process. In this context and under conditions of normality, this paper presents two procedures, computational and analytical, for estimating the average gene expression of GM maize, providing examples to show the application of the proposed methods. Additionally, through simulation we reproduced several circumstances that a researcher might find, providing a computational algorithm in the statistical program R (R Development Core Team 2007) to create other scenarios. Both procedures ensure that, W confidence interval amplitude is lower or equal to the desired amplitude ω with γ probability. This is achieved because both methods consider the randomness of the sampling variance. However, we recommend the use of the formula proposed and analytical procedure, because it is accurate and easy to use.
Key words: confidence interval, average gene expression, normality.
Introducción
La diseminación de transgenes en los cultivos normales o parientes silvestres es un riesgo inherente en la agricultura. En México, un país que alberga más 60% de la variedad genética del maíz (Zea mays L.) (Piñeyro-Nelson et al., 2009), los efectos por diseminar genes de plantas de maíz genéticamente modificadas (GM), son desconocidos, aún cuando investigaciones recientes testifican la presencia de transgenes de maíz en las regiones sur-este y centro-oeste del país (Dyer et al., 2009; Piñeyro-Nelson et al., 2009). Ante este escenario de preocupación es importante detectar la presencia y expresión de trangenes en los cultivos de maíz.
La detección de plantas transgénicas involucra la evaluación de compuestos orgánicos o el análisis del metabolismo de cada uno de los elementos de la población. Sin embargo, efectuar una inspección o análisis a cada elemento consumiría grandes cantidades de recursos económicos y materiales. Una alternativa para disminuir costos es efectuar muestreo en grupos antes de realizar las mediciones analíticas, esto reduciría el total de análisis. Además, usar muestreo en grupos para caracterizar poblaciones no solo es más eficiente en términos económicos, sino también pueden realizarse estimaciones más precisas y menos sesgadas que las obtenidas con muestras individuales (Caudill, 2010).
El muestreo en grupos o prueba de grupos, propuesto por Dorfman (1943), consiste en formar g grupos de tamaño k con muestras individuales. Esta forma de agrupar elementos puede usarse para: a) reducir la variación biológica; b) disminuir costos vía la reducción del total de pruebas de laboratorio; y c) que la disponibilidad de muestras limitadas no sea un problema. Debido al ahorro significativo de tiempo y dinero por utilizar esta técnica, su empleo se incrementa día a día, usándose para detectar enfermedades al donar sangre (Dodd et al., 2002), drogas (Remlinger et al., 2006), plantas transgénicas (Hernández-Suárez et al., 2008; Montesinos-López et al., 2010), estimar la prevalencia de enfermedades humanas (Verstraeten et al., 1998), enfermedades de plantas (Tebbs y Bilder, 2004) y animales (Peck, 2006).
En la prueba de grupos, grupos de individuos son caracterizados en lugar de elementos individuales. De acuerdo con el supuesto del promedio biológico, la medición en una muestra en grupo es comparable con la media aritmética de los niveles individuales de los elementos que conforman el mismo grupo (Mary-Huard et al., 2007; Caudill, 2010). Por lo tanto, si una cantidad medible se distribuye normalmente, la distribución de las medidas del muestreo en grupos también tendrán distribución normal con la misma media pero con varianza reducida proporcionalmente por el número de elementos en el grupo.
Cuando el objetivo es estimar la expresión media de un gen que produce plantas GM en la población, es importante diseñar un experimento que garantice el tamaño de muestra apropiado para asegurar intervalos de confianza cortos (Shaarschimidt, 2007). Un tamaño de muestra pequeño no garantiza buena precisión en la estimación del parámetro de interés, mientras un tamaño de muestra grande es un derroche innecesario de recursos (Wang et al., 2005). En el contexto de los experimentos de microarreglos y considerando variables aleatorias continuas, Kendziorski et al. (2003), Dobbin y Simon (2005) y Zhang y Gant (2005) determinaron los tamaños de muestra bajo el muestreo en grupos. Kendziorski et al. (2003) proporcionó una fórmula que determina el número de grupos para estimar la expresión de genes y establecer intervalos de confianza (ICs), comparando los resultados con aquellos obtenidos sin utilizar prueba en grupos. Sin embargo, el trabajo ignora la naturaleza estocástica de la amplitud del intervalo de confianza (IC).
Por otro lado, Dobbin y Simon (2005) y Zhang y Gant (2005) obtuvieron el tamaño de muestra bajo el enfoque de potencia en muestreo en grupos, razón por la cual los resultados no son apropiados para producir estimaciones precisas de la media u otro parámetro de interés.
Típicamente se han propuesto fórmulas para determinar tamaños de muestra bajo condiciones de potencia. Este enfoque es adecuado cuando se hacen pruebas de hipótesis, reportando los resultados en términos de p-values. Sin embargo, para hacer inferencias actualmente se ha incrementado el uso de ICs en lugar de pruebas de hipótesis (Pan y Kupper, 1999). En los estudios agrícolas la estimación de parámetros vía IC es importante porque frecuentemente el objetivo principal es estimar la magnitud del efecto de interés, y no sólo decidir si los efectos de los tratamientos son estadísticamente diferentes.
Una prueba de hipótesis puntualiza si un efecto es significativo sin proporcionar una caracterización precisa del efecto que está siendo probado en la hipótesis nula. El uso de ICs asegura no solo que la magnitud del efecto pueda evaluarse, sino también que el efecto en estudio pueda ser identificado fácilmente por el lector. Además, los ICs transmiten información para que la magnitud del efecto pueda determinarse a partir de los datos disponibles (Beal, 1989). Por tales razones se ha puesto atención al calcular tamaños de muestras apropiados para realizar inferencias basadas en ICs. Este enfoque de estimación se ha denominado precisión en la estimación de parámetros (PEP) porque cuando la amplitud del IC con una confiabilidad de (1 - α) 100% decrece, la precisión esperada de la estimación aumenta (Kelley y Maxwell, 2003; Kelley y Rausch, 2006; Kelley, 2007).
Cuando se determina el tamaño de muestra se requieren los valores de algunos parámetros. En la práctica éstos son desconocidos y usualmente se estiman de la literatura o estudios previos. Éstas estimaciones son consideradas como los verdaderos valores de los parámetros, trayendo como consecuencia que con el tamaño de muestra calculado no se logre la precisión deseada en el IC (Wang et al., 2005). Para tener en cuenta la incertidumbre inducida por el error de muestreo, Kelley (2007) y Kupper y Hafner (1989) señalaron que la naturaleza estocástica de la amplitud del IC debería considerarse para no subestimar el tamaño de muestra requerido. Así, bajo el modelo de Dorfman, Montesinos-López et al. (2010) propuso un procedimiento que determina el tamaño de muestra para estimar la proporción de plantas transgénicas, asegurando que la amplitud W del IC sea más estrecha que el valor deseado ω. Sin embargo, este método no proporciona una solución analítica.
Por tales motivos, bajo el contexto de la prueba de grupos con prueba perfecta y tamaño de grupo fijo, el objetivo de esta investigación fue proponer un método analítico que determine el tamaño de muestra dado en términos del número de grupos requeridos para estimar por intervalo la expresión media de un gen, asegurando ICs estrechos. La precisión en la estimación de la media se logra porque se considera la aleatoriedad de la amplitud del IC. Además se presenta un algoritmo computacional en el programa estadístico de uso libre y distribución gratuita R (R Development Core Team 2007) para obtener los resultados, de tal forma que los investigadores puedan reproducir otros escenarios.
Materiales y métodos
Sea X la cantidad medida que está siendo determinada en la población o experimento; es decir, el nivel de expresión de un gen. Permita que xi denote el valor de la variable X en el elemento i de la población de interés. Se asume que todas las xis en la población son independientes, normalmente distribuidas con media µ y varianza σ2, denotado por xi ∼ N(µ, σ2), para toda i.
Los elementos de la población son seleccionados aleatoriamente y a cada uno se le extrae una muestra de tejido. Un grupo de muestras de tejidos es formado congregando k muestras de tejidos de elementos individuales, los cuales son seleccionados aleatoriamente (Zhang y Gant, 2005). Así se forman g grupos de k elementos cada uno, donde g, k son enteros positivos y n= g, k. n es el número total de muestras individuales (o sujetos), aunque en este caso no se realizan las mediciones de las muestras individuales. En su lugar, estas muestras individuales son agrupadas en g grupos con k muestras en cada grupo y m mediciones (réplicas) se efectúan en cada grupo de muestras de tejidos. Por lo tanto, m es el número de réplicas técnicas de la medida en cada grupo (Zhang y Gant, 2005; Caudill, 2010). Note que si k =1, el experimento es equivalente a no realizar grupos de muestras de tejidos; y si m=1 , no existen réplicas.
Bajo el supuesto básico del promedio biológico, el resultado por agrupar k muestras de tejidos en proporciones iguales es que el valor de k en cada grupo es el promedio de los elementos que conforman este mismo grupo (Zhang y Gant, 2005), Además, x= k-1 Σki=1 xi tiene distribución normal con media µ y varianza k-1σ2 para cada grupo de la población (Zhang y Gant, 2005). En este artículo sólo se discuten muestras de grupos con contribuciones individuales iguales. Aunque se pueden formar grupos con contribuciones desiguales de las muestras individuales, tal diseño es generalmente menos efectivo que con contribuciones iguales (Peng et al., 2003).
De acuerdo con Zhang y Gant (2005), cuando se toma una medida sobre un grupo p, el valor medido es yp,r=  p + εr, donde p indica el grupo, r hace referencia a las mediciones y εr son errores aleatorios, los cuales se asume son independientes con distribución normal εr ∼ N(0,σ2ε). De aquí en adelante a σ2ε se le llamará la varianza técnica y σ2 la varianza biológica de la población. Las mediciones sobre los g grupos son los resultados del experimento. Así se tiene yp,r‚ para p= 1,..., g, r= 1,..., m; y g es el número de grupos formados a partir de una muestra de la población (Zhang y Gant, 2005). El propósito es realizar inferencias sobre las propiedades de la población con base en los datos disponibles.
p + εr, donde p indica el grupo, r hace referencia a las mediciones y εr son errores aleatorios, los cuales se asume son independientes con distribución normal εr ∼ N(0,σ2ε). De aquí en adelante a σ2ε se le llamará la varianza técnica y σ2 la varianza biológica de la población. Las mediciones sobre los g grupos son los resultados del experimento. Así se tiene yp,r‚ para p= 1,..., g, r= 1,..., m; y g es el número de grupos formados a partir de una muestra de la población (Zhang y Gant, 2005). El propósito es realizar inferencias sobre las propiedades de la población con base en los datos disponibles.
Puede mostrarse que Ῡ= (mg)-1Σgp=1Σmr=1 yp,r es un estimador insesgado de µ (Zhang y Gant, 2005; Caudill, 2010), con varianza σῩ2= g-1(k-1σ2 + m-1σ2∈)= g-1σ2p, donde σ2p= k-1σ2 + m-1σ2 ∈, y sῩ2= g-1((g - 1)-1 Σ gp=1(m-1Σmr=1 yp,r -Ῡ)2)= g-1σ2p es un estimador insesgado de σῩ2 (Zhang y Gant, 2005; Kendziorski et al., 2003), donde sp2= (g - 1)-1 Σ gp=1(m-1Σmr=1 yp,r -Ῡ)2. Por lo tanto, el correspondiente IC de Wald es:

Donde: Z1-α/2 es el cuantil 1-α/2 de la distribución normal estándar. La ecuación (1) es igual a la propuesta por Kendziorski et al. (2003), pero con la diferencia de que se usa Z1-α/2 en lugar del cuantil 1-α/2 de la distribución t-student con g-1 grados de libertad. Esta sustitución no provoca problemas graves de subestimación. Por otro lado, este IC es fácil de calcular y permite derivar una fórmula cerrada para el tamaño de muestra, aunque cuando g es pequeño, la amplitud del IC es más grande.
Derivación del tamaño de muestra para estimar la media
La cantidad  en la ecuación (1), la cual es sumada y sustraída a la media observada ӯ, se define como W/2 (W es la amplitud total del IC). Los límites superior e inferior del IC están determinados por W/2. El grado de precisión del IC es el valor de más interés en trabajos con enfoques PEP. El valor W se fija a priori de acuerdo a la precisión deseada al estimar los parámetros.
en la ecuación (1), la cual es sumada y sustraída a la media observada ӯ, se define como W/2 (W es la amplitud total del IC). Los límites superior e inferior del IC están determinados por W/2. El grado de precisión del IC es el valor de más interés en trabajos con enfoques PEP. El valor W se fija a priori de acuerdo a la precisión deseada al estimar los parámetros.
La amplitud total del IC [Ecuación (1)] puede expresarse como  . Para estimar el número de grupos necesarios (tamaño de muestra) con el fin de estimar la media µ, dada una amplitud esperada ω (error), debe resolverse la Ecuación
. Para estimar el número de grupos necesarios (tamaño de muestra) con el fin de estimar la media µ, dada una amplitud esperada ω (error), debe resolverse la Ecuación  en términos de g(haciendo W= ω), y la varianza muestral sp2 debe reemplazarse por el valor poblacional σ2p, produciéndose así la siguiente fórmula:
en términos de g(haciendo W= ω), y la varianza muestral sp2 debe reemplazarse por el valor poblacional σ2p, produciéndose así la siguiente fórmula:

Ésta fórmula puede usarse para estimar el número de grupos requeridos que se usaran en la estimación de la expresión media de un gen, considerando un tamaño de grupo fijo k, m mediciones por grupo, y asumiendo que σ2 y σ2 ε son conocidas. Note que si k= m= 1, la Ecuación (2) se reduce a la fórmula estándar para estimar la media bajo muestreo aleatorio simple [n= w-24Z21-α/2σ2]. Sin embargo, en la ecuación (2) los valores de σ2 y σ2 ε son desconocidos, por lo que se usan sus respectivos estimadores.
Con la ecuación (2) se determina el tamaño de muestra que arroje un IC de amplitud W para estimar la media, bajo el contexto de la prueba en grupos. Sin embargo, no existe garantía de que la amplitud observada W sea precisa para algún IC en particular porque se usan estimaciones de σ2 y σ2ε. Esto implica que aproximadamente 50% de la distribución muestral de W sea menor que ω (Kelley y Maxwell, 2003; Kelley et al., 2003; Montesinos-López et al., 2010). En seguida se propone una fórmula para calcular tamaños de muestra óptimos que garantizan ICs suficientemente precisos.
Procedimiento para calcular tamaños de muestra óptimos
La amplitud del IC para la media es 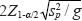 , donde sp2= (g -1)-1 Σgp=1 (m-1Σmr=1yp,r - Ῡ)2 W debería ser menor que un valor especifico ω con probabilidad γ. Así, siguiendo la lógica de Hahn y Meeker (1991) y Montesinos-López et al. (2011), para determinar un tamaño de muestra que asegure ICs estrechos, el tamaño de muestra óptimo es el valor entero más pequeño gm tal que:
, donde sp2= (g -1)-1 Σgp=1 (m-1Σmr=1yp,r - Ῡ)2 W debería ser menor que un valor especifico ω con probabilidad γ. Así, siguiendo la lógica de Hahn y Meeker (1991) y Montesinos-López et al. (2011), para determinar un tamaño de muestra que asegure ICs estrechos, el tamaño de muestra óptimo es el valor entero más pequeño gm tal que:

Por lo tanto, el número de grupos requeridos es

Donde: γ es el grado de certeza (probabilidad requerida) para lograr que la amplitud observada del IC W no sea mayor que el valor deseado ω; X2gm-1,γ es el cuantil γ de la distribución chi-cuadrada con gm - 1 grados de libertad. Con la ecuación (3) se obtiene el tamaño de muestra requerido gm, asegurando que el IC W sea menor o igual a la amplitud deseada ω con una probabilidad de al menos γ. Note que si el nivel de certeza deseado es γ= 0.5, la cantidad X2gm-1,γ / (gm - 1) es aproximadamente igual a 1, por lo que la ecuación (3) se reduce a la ecuación (2), aunque la ecuación (3) considera la aleatoriedad de los estimadores de σ2 y σ2 ε vía el grado de certeza deseado γ. Sin embargo, un inconveniente para derivar el tamaño de muestra exacto es que gm esta en ambos lados de la ecuación (3), requiriéndose de un procedimiento iterativo para resolver la ecuación en términos de gm.
Aproximación del tamaño de muestra óptimo
Si se usa gm= g [obtenido en la ecuación (2)] en el lado derecho de la ecuación (3), se tiene una solución analítica. Esto implica que gm sea igual a:

Donde: X2g-1,γ es el cuantil γ de la distribución chi-cuadrada con g - 1 grados de libertad y g es el tamaño de muestra obtenido con la ecuación (2).
Resultados y discusión
Usando el programa R (R Development Core Team, 2007), en el apéndice se proporciona información para implementar los métodos propuestos y así obtener los tamaños de muestra para cualquier combinación de σ2, σ2ε, m, ω, γ y α. Los valores del Cuadro 1 fueron calculados con el método exacto [ecuación (3)]. Estos valores están basados en resultados para detectar y estimar la expresión media de un gen.
Tamaño de muestra, cuadro 1
Suponga que un investigador está interesado en estimar la expresión media de un gen de maíz GM, en la región de Oaxaca, México, donde Quist y Chapela (2001) reportaron el hallazgo de transgenes de maíz. Con esta información y después de revisar estudios previos, se hipotetiza que la varianza biológica es σ2= 0.1, la varianza técnica es σ2ε= 0.02, con IC de 95%, tamaño de grupos k=10, réplicas técnicas igual m=2, y se desea que la amplitud observada del IC sea menor o igual a 0.05, es decir Wx= (µU - µL) < ω= 0.05. La aplicación del método exacto señala que se requiere una muestra preliminar de g= 123 grupos, cada uno de tamaño k=10. Este tamaño de muestra se encuentra en el primer sub-cuadro del Cuadro 1, con γ= 0.5, k=10, σ2= 0.1, σ2ε= 0.02 y ω= 0.05.
Sabiendo que los g= 123 grupos producirán ICs precisos sólo 50% de las veces, el investigador incorpora a la estimación una certeza de γ= 0.99, lo cual implica que la amplitud de 95% de confiabilidad del IC sea mayor a la amplitud requerida ω= 0.05 no más de 1% de las veces. En el tercer sub-cuadro del Cuadro 1 (gm con γ= 0.99) se observa que el tamaño de muestra con el procedimiento modificado arroja gm= 158 grupos. Por lo tanto, usando 158 grupos se tendrá una certeza de 99% de que la amplitud observada W del IC no será más grande que ω= 0.05 al estimar la media µ. Este tamaño de muestra se localiza en el tercer sub-cuadro del Cuadro 1 (gm con γ= 0.99, k=10, σ2= 0.1, σ2ε= 0.02 y ω= 0.05). El uso del Cuadro 2 es similar, con la diferencia de que éste contiene diferentes valores para la amplitud deseada (ω) y sólo un valor para la varianza técnica (σ2ε= 0.0125).
Comparación de los métodos exacto y aproximado usando grupos de tamaño k= 5
Con k= 5, la ecuación (4) arroja casi los mismos resultados que se obtienen con el método exacto [ecuación (3) y considerando γ= 0.5]. Sin embargo, si γ= 0.9, las diferencias en el número de grupos entre los dos métodos son uno o dos grupos; en este caso el método aproximado produce una ligera sobreestimación. También, si γ= 0.99, el método aproximado produce entre cuatro y seis grupos más que los calculados con el método exacto. Esto indica que si γ= 0.99, la diferencia entre los dos enfoques se incrementa, en tal caso la fórmula analítica sobreestima ligeramente el número óptimo de grupos.
Por otro lado, usando muestras de tamaño k= 5 (Cuadro 4), si γ= 0.5, el número de grupos requeridos con ambos métodos es el mismo. Si γ= 0.9, el método aproximado requiere entre uno y dos grupos más que el método exacto. Sin embargo, si γ= 0.99, el método aproximado necesita entre cinco y seis grupos más que el método exacto, indicando una ligera sobreestimación del número óptimo de grupos, igual a lo ocurrido con grupos de tamaño k=5. Pero la ventaja del método aproximado [ecuación (4)] es que tiene solución analítica, la cual es una fórmula cerrada muy simple.
Tamaño de muestra óptimo- ejemplo usando la fórmula propuesta
Suponga que un investigador está interesado en estimar la expresión media de un gen de plantas GM y no tiene acceso a los Cuadros 2 y 3, ni al paquete R. El investigador hipotetiza que la varianza biológica y la varianza técnica son σ2= 0.1 y σ2ε= 0.02, respectivamente. Además, el IC es de 95% (Z1-0.05/2= 1.96), el tamaño del grupo es k=10, las réplicas técnicas son m=2, y se desea que la amplitud final del IC sea menor o igual a 0.05, es decir Wx= (µU - µL) < ω= 0.05. Primero se calcula el tamaño de muestra inicial con la ecuación (2):

Es decir, se requieren g= 123 grupos, los cuales conllevarán a ICs con la precisión deseada sólo 50% de las veces. Por esta razón, el investigador incorpora una certeza de γ= 0.99, para lo cual usa la ecuación (4), obteniendo así el tamaño de muestra modificado que le permitirá lograr sus objetivos. Como los resultados indican que la muestra preliminar es g= 123 y el cuantil γ= 0.99 de la distribución chi-cuadrada con g - 1= 122 grados de libertad, es X2122,0.99= 161.2495, el tamaño de muestra modificado es gm= g(X2g-1,γ) / (g - 1)= 123(161.2495) / 122= 163. Esto significa que el número de grupos requeridos es 163, mientras el método exacto necesita 158, cinco grupos menos que el método aproximado. Note que la ecuación (4) produce una ligera sobreestimación pero con la ventaja de que puede determinarse fácilmente.
En general, dentro de un rango especifico para k y γ, los resultados de la fórmula fueron muy precisos, aunque la fórmula propuesta sobreestima el número óptimo de grupos, principalmente para γ > 0.99. Es importante señalar que la fórmula derivada asume normalidad de los datos. Por lo tanto, antes de aplicar la expresión propuesta se necesita corroborar este supuesto. Además, los métodos presentados asumen sensibilidad y especificidad perfecta, los cuales deben considerarse al diseñar el estudio.
Conclusiones
Los cuadros proporcionan los tamaños de muestra pertinentes de una amplia variedad de escenarios para estimar la expresión media de un gen, garantizando ICs precisos. Sin embargo, los resultados no cubren todas las combinaciones de k, σ2, σ2 ε, m, ω, γ, y α; por lo que se recomienda usar la fórmula del tamaño de muestra óptimo [ecuación (3)] y el programa desarrollado en el paquete R (R Development Core Team, 2007) ya que permite a los usuarios determinar el tamaño de muestra de manera fácil y rápida de acuerdo a los requerimientos o necesidades. No obstante, si el investigador no tiene acceso al programa R, la solución práctica es usar la ecuación (4). Esta fórmula analítica [ecuación (4)] tiene la ventaja sobre el método computacional exacto [ecuación (3)] porque no necesita del programa R para obtener tamaños de muestra apropiados.
Además, es preferible usar la fórmula analítica que el método estándar [ecuación (2)] puesto que este último arroja tamaños de muestra más pequeños, los cuales producirán probabilidades muy bajas de lograr los objetivos inferenciales (típicamente menor que 0.5). Se recomienda usar la fórmula analítica con tamaños de grupo menores o iguales a 25. Esta recomendación es análoga con las expuestas en varios estudios cuando se usan pruebas de grupo con variables aleatorias continuas (Kendziorski et al., 2003; Dobbin y Simon, 2005; Zhang y Gant, 2005).
Literatura citada
Beal, S. L. 1989. Sample size determination for confidence intervals on the population mean and on the difference between two population means. Biometrics. 45(3):969-977. [ Links ]
Caudill, S. P. 2010. Characterizing populations of individuals using pooled samples characterization. J. Exp. Sci. Environ. Epidemiol. 20(1):29-37. [ Links ]
Dobbin, K. and Simon, R. 2005. Sample size determination in microarray experiments for class comparison and prognostic classification. Biostatistics. 6(1):27-38. [ Links ]
Dodd, R.; Notari, E. and Stramer, S. 2002. Current prevalence and incidence of infectious disease markers and estimated window-period risk in the American Red Cross donor population. Transfusion. 42(8):975-979. [ Links ]
Dorfman, R. 1943. The detection of defective members of large populations. The Annals of Mathematical Statistics. 14(4):436-440. [ Links ]
Dyer, G. A.; Serratos-Hernández, J. A.; Perales, H. R.; Gepts, P.; Piñeyro-Nelson, A.; Chávez, A.; Salinas-Arreorta, N.; Yúnez-Naude, A.; Taylor, J. E. and Álvarez-Buylla, E. R. 2009. Dispersal of transgenes through maize seed systems in Mexico. PLoS ONE. 4(5):e5734. [ Links ]
Hahn, G. J. and Meeker, W. A. 1991. Statistical intervals: a guide for practitioners. Hoboken, NJ: John Wiley and Sons, Inc. 392 p. [ Links ]
Hernández-Suárez, C. M.; Montesinos-López, O. A.; McLaren, G. and Crossa, J. 2008. Probability models for detecting transgenic plants. Seed Sci. Res. 18(2):77-89. [ Links ]
Kelley, K. 2007. Sample size planning for the coefficient of variation from the accuracy in parameter estimation approach. Behavior Res. Methods. 39(4):755-766. [ Links ]
Kelley, K. and Maxwell, S. E. 2003. Sample size for multiple regression: obtaining regression coefficients that are accurate, not simply significant. Psychol. Methods. 8(3):305-321. [ Links ]
Kelley, K. and Rausch, J. R. 2006. Sample size planning for the standardized mean difference: Accuracy in parameter estimation via narrow confidence intervals. Psychol. Methods. 11(4):363-385. [ Links ]
Kelley, K.; Maxwell, S. E. and Rausch, J. R. 2003. Obtaining power or obtaining precision: delineating methods of sample size planning. Eval. Health Profess. 26(3):258-287. [ Links ]
Kendziorski, C. M.; Zhang, Y.; Lan, H. and Attie, A. 2003. The efficiency of pooling mRNA in microarray experiments. Biostatistics. 4(3):465-477. [ Links ]
Kupper, L. L. and Hafner, K. B. 1989. How appropriate are popular sample size formulas? The American Statistician. 43(2):101-105. [ Links ]
Mary-Huard, T.; Daudin, J.; Baccinim.; Biggeri, A. and Bar-Hen A. 2007. Biases induced by pooling samples in microarray experiments. Bioinformatics. 23(13):i313-i318. [ Links ]
Montesinos-López, O. A.; Montesinos-López, A.; Crossa, J.; Eskridge, K. and Hernández-Suárez, C. M. 2010. Sample size for detecting and estimating the proportion of transgenic plants with narrow confidence intervals. Seed Sci. Res. 20(2):123-136. [ Links ]
Montesinos-López, O. A.; Montesinos-López, A.; Crossa, J.; Eskridge, K. and Sáenz, R. A. 2011. Optimal sample size for estimating the proportion of transgenic plants using the Dorfman model with a random confidence interval. Seed Sci. Res. 21(3):235-246. [ Links ]
Pan, Z. and Kupper, L. 1999. Sample size determination for multiple comparison studies treating confidence interval width as random. Statistics in Medicine. 18(12):1475-1488. [ Links ]
Peck, C. 2006. Going after BVD. Beef. 42:34-44. [ Links ]
Peng X.; Wood, C. L.; Blalock, E. M.; Chen, K. C.; Landfield, P. W. and Stroberg, A. J. 2003. Statistical implications of pooling RNA samples for microarray experiments. BMC Bioinformatics. 4:26. [ Links ]
Piñeyro-Nelson, A.; Van Heerwaarden, J.; Perales, H. R.; Serratos-Hernández, J. A. and Rangel, A. 2009. Transgenes in Mexican maize: molecular evidence and methodological considerations for GMO detection in landrace populations. Mol. Ecol. 18(4):750-761. [ Links ]
Quist, D. and Chapela, I. H. 2001. Transgenic DNA introgressed into traditional maize landraces in Oaxaca, Mexico. Nature. 414:541-543. [ Links ]
R Development Core Team. 2007. R: a language and environment for statistical computing [Computer software and manual], R Foundation for Statistical Computing. URL: http://www.r-project.org. [ Links ]
Remlinger, K.; Hughes-Oliver, J.; Young, S. and Lam, R. 2006. Statistical design of pools using optimal coverage and minimal collision. Technometrics. 48(1):133-143. [ Links ]
Schaarschmidt, F. 2007. Experimental design for one-sided confidence interval or hypothesis tests in binomial group testing. Communications in biometry and Crop Science. 2(1):32-40. [ Links ]
Tebbs, J. M. and Bilder, C. R. 2004. Confidence interval procedures for the probability of disease transmission in multiple-vector-transfer designs. J. Agric. Biol. Environ. Statistics. 9(1):75-90. [ Links ]
Verstraeten, T.; Farah, B.; Duchateau, L. and Matu, R. 1998. Pooling sera to reduce the cost of HIV surveillance: a feasibility study in a rural Kenyan district. Tropical Medicine and International Health. 3(9):747-750. [ Links ]
Wang, H.; Chow, S.C. and Chen, M. 2005. A Bayesian approach on sample size calculation for comparing means. J. Biopharmaceutical Statistics. 15(5):799-807. [ Links ]
Zhang, S. D. and Gant, T. W. 2005. Effect of pooling samples on the efficiency of comparative studies using microarrays. Bioinformatics. 21(24):4378-4383. [ Links ]

