










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkNova scientia
versión On-line ISSN 2007-0705
Nova scientia vol.5 no.10 León oct. 2013
Ciencias naturales e ingenierías
Formalismo de cálculo de la movilidad de portadores en un pozo cuántico delta dopado (δ -DQW)
Electric Charge Mobility in a delta-doped quantum well (δ-DQW)
H. Rodriguez-Coppola1, M. E. Mora-Ramos2y L. M. Gaggero-Sager2
1 Departamento de Física Aplicada, Facultad de Física, Universidad de La Habana, Cuba.
2 Facultad de Ciencias, Universidad Autónoma del Estado de Morelos, México.
Huberto Rodriguez-Coppola. E-mail. huberto.rodriguezcoppola@gmail.com.
Recepción: 14-10-2012
Aceptación: 18-02-2013
Resumen
Este artículo ofrece un esquema para el cálculo de la movilidad de electrones en una estructura de pozo delta dopado, a bajas temperaturas y para campos eléctricos aplicados no muy intensos. El análisis se hace para un sistema dopado tipo n, considerando una lámina de impurezas de silicio en una muestra de GaAs. Se tiene en cuanta explícitamente el carácter tridimensional de los estados electrónicos y de las magnitudes que se miden -en lugar de realizar aproximaciones en que se reduzcan las dimensiones del sistema-, lo que facilita la ejecución del cálculo. Se exponen los resultados de otros esquemas utilizados y se dan las fórmulas que se emplearán para comprobar y comparar nuestros cálculos con los reportados previamente. En la actualidad, se está en el proceso de implementación del cálculo numérico, del que se expone el algoritmo que se tiene en ejecución.
Palabras Clave: Pozo delta dopado; Nanoestructura semiconductora; Movilidad eléctrica; Campo eléctrico.
Abstract
This paper offers a scheme for the calculation of the mobility of electrons in a delta doped QW considering the conditions of low temperature and low electric field intensity. The system considered is a quantum well for electrons in the conduction band made by a silicon impurity plane in a sample of GaAs. The 3D character of the electronic states and of the magnitudes considered is explicitly tacked into account instead of reducing the dimensionality of the system to make the calculations more easy. Other calculations are compared and the formulae obtained in our approximation are given. At this moment the numerical calculation is in progress and the algorithm to do it is presented.
Keywords: delta doped QW; electric charge mobility; electric field.
1. Introducción
Un elemento importante para el conocimiento y la predicción del comportamiento de las magnitudes de transporte de carga de un sistema semiconductor, es la movilidad de los portadores, definida como la respuesta de estos a la acción del campo eléctrico aplicado, que resulta en el movimiento. Desde hace alrededor de dos décadas se comenzó el estudio de la movilidad de electrones en sistemas delta dopados (puede consultarse, por ejemplo, González 1994,1996, 1999; Shi 1997; Hai 1995; Hai et al. 1995) como un problema de interés práctico, buscando elevar la velocidad de movimiento de los portadores en el sistema y así mejorar la respuesta de estos a campos eléctricos externos aplicados. Con esto se podría implementar su uso en dispositivos electrónicos de respuesta rápida. Además, el tratamiento dado tiene utilidad como elemento teórico de relevancia para comprender la forma en que se mueven y se comportan los portadores de carga en estos sistemas y mejorar sus características de uso.
La dificultad en el estudio de la movilidad de los electrones en un sistema delta-dopado tipo n de Si en GaAs proviene del hecho que es la propia distribución de átomos de Si ionizados la que condiciona la forma del pozo de potencial en que se confinan estos portadores de carga, con varias subbandas ocupadas a bajas temperaturas. Esto ha tornado dicho análisis en un asunto complicado, tanto en la banda de conducción (BC) como en la banda de valencia (BV). En los reportes (Gaggero 2002; Gaggero 1998), se investiga la forma en que se pueden obtener los niveles de energía en un pozo cuántico delta-dopado, sin recurrir al proceso autoconsistente que implica la solución simultánea de las ecuaciones de Schrödinger y Poisson para reconstruir el potencial y, de esa forma, calcular los estados electrónicos del movimiento en el potencial de las impurezas ionizadas. El esquema estudiado por Gaggero et al. fue inicialmente propuesto por Ioriatti (Ioriatti 1990) y su significación estriba en que reduce el cálculo de los estados energéticos y las funciones de onda de este sistema a un problema relativamente simple, en el que se usa una expresión analítica para describir el potencial del pozo. El proceso de determinación de la movilidad electrónica en esta clase de sistemas se ha desarrollado desde los finales de los años 80 y tuvo su expresión más depurada en los trabajos (González 1994; González 1996).
El completamiento de la tarea de evaluar la movilidad de portadores se compone de varias partes importantes. En el caso específico de un pozo delta-dopado podemos delinear los siguientes pasos: i) Calcular los estados energéticos y las funciones de onda correspondientes a esos estados.
Esto se puede hacer utilizando o no el esquema autoconsistente. ii) Efectuar el cálculo de los elementos matriciales de la interacción de los electrones con los operadores de interacción asociados a los diferentes mecanismos que provocan dispersión en el material (impurezas ionizadas, impurezas neutras, fonones de diferentes ramas de oscilación, etc.). Previo a este paso, se debe discutir la importancia relativa de cada una de estas interacciones, seleccionando los mecanismos más importantes de acuerdo con las condiciones particulares de operación del sistema electrónico de baja dimensión. iii) Dentro del problema de la interacción entre los portadores y los átomos de impurezas, especialmente, habrá que tener en cuenta el apantallamiento del potencial coulombiano de las impurezas asociado al propio gas electrónico. Esto se puede realizar en el marco de diferentes aproximaciones, de acuerdo principalmente a la forma en que se incluyen los efectos de muchos cuerpos en el sistema. iv) Determinar la movilidad de los electrones en la estructura. Una de las vías más directas de realizar este cálculo es plantear y resolver la ecuación de Boltzmann para el movimiento de los electrones en la aproximación del tiempo de relajación. Esta última cantidad es proporcional a la movilidad de los portadores en las condiciones en que fue resuelta la mencionada ecuación de transporte. En (González 1994,1999; Hai et al. 1995) se describe este procedimiento para un sistema delta-dopado.
En términos generales, los resultados contenidos en los trabajos desarrollados por los referidos autores siguen la tendencia de los reportados experimentalmente; pero adolecen de determinados defectos que pueden resumirse en lo siguiente: i) Se obtienen saltos en los valores de la movilidad cuando se pasa de una concentración de impurezas a otras, lo que no se observa en los experimentos (González 1994; Hai 1995). ii) Los valores de la movilidad que se reportan a partir del cálculo teórico son muy diferentes (en ocasiones órdenes de magnitud) a los obtenidos en los experimentos (González. 1994). iii) Los cálculos que se realizan, aunque incluyen varias subbandas ocupadas, no consideran el movimiento de los electrones en tres dimensiones. En otras palabras, reducen la descripción de los estados a un problema estrictamente bidimensional y no en el espacio (González 1994). Esto tiene consecuencias tanto en la descripción de los estados como en las magnitudes que se calculan.
El presente trabajo describe el proceso seguido por nosotros para obtener de forma teórica la movilidad de electrones en un pozo cuántico delta-dopado. Hacemos especial énfasis en aquellos elementos que diferencian el enfoque que proponemos con el desarrollado -en el mismo esquema del tiempo de relajación- por los autores mencionados arriba, y exponemos sus particularidades con un elevado grado de detalle. Se hace de esta manera porque los aspectos generales de dicho enfoque son aplicables a una gran mayoría de los sistemas electrónicos cuasibidimensionales. Un elemento clave en el formalismo es la evaluación de la función dieléctrica del gas tridimensional de portadores con confinamiento en una dirección espacial, la cual contiene la información del apantallamiento de la interacción coulombiana entre todos los electrones del gas. En el caso de los sistemas delta-dopados se han obtenido ya algunos resultados en ese tema (Rodríguez-Coppola 2010).
Es preciso hacer notar que, dada la extensión del trabajo en su conjunto, hemos decidido presentar en este reporte únicamente la parte concerniente al formalismo. Dejamos para una publicación posterior la presentación de los resultados específicos para la movilidad de electrones en pozos cuánticos delta-dopados tipo n en GaAs, la cual se estudiará fundamentalmente como función de la concentración de átomos de impureza ionizados en el sistema.
Para el desarrollo de este propósito se ha dividido el texto en las siguientes partes: la sección siguiente se dedica a describir brevemente el proceso de cálculo de los estados electrónicos del sistema delta dopado en las condiciones planteadas, utilizando la aproximación de Thomas-Fermi (Gaggero 2002). Los resultados de esta evaluación, para el conjunto de pozos a modelar, se utilizan como datos iniciales para el cálculo de la interacción de los electrones con los procesos de dispersión presentes en el sistema. Las consideraciones para la selección de los procesos de dispersión y la forma en que se incluyen en el cálculo los diferentes procesos, se describe en la siguiente sección. Posteriormente, se describe el proceso de obtención de los tiempos de relajación y su relación con la movilidad de los portadores y se dan las consideraciones finales sobre el trabajo realizado.
2. Cálculo de los estados electrónicos de un pozo delta dopado
El procedimiento más exacto para determinar los estados electrónicos de un δ-DQW requiere del proceso autoconsistente de solución de las ecuaciones diferenciales de Schrödinger y de Poisson porque el propio potencial asociado a las impurezas ionizadas es el que produce la deformación de la banda de conducción que constituye el pozo de energía potencial que confina a los electrones en la vecindad de la capa donde están dispuestas las impurezas (supuesta idealmente una monocapa y, en la práctica, resultando normalmente unas pocas capas atómicas insertadas en la matriz del semiconductor volumétrico). En la literatura se han realizado diferentes alternativas para el análisis de esta situación que tratan de sustituir el tratamiento autoconsistente por alguna aproximación que desde el punto de vista cuantitativo conduzca a desviaciones importantes de los valores de los niveles de energía con respecto a los proporcionados por el sistema de Schrödinger-Poisson. De entre las propuestas nos va a interesar aqui la que permite deducir una expresión analítica para el perfil de potencial asociado al δ-DQW, la cual se deriva de la aplicación de la aproximación de Thomas-Fermi al problema de densidad funcional local en una dimensión (Ioriatti 1990; Gaggero, 2002; Gaggero 1998).
Debemos comenzar diciendo que consideraremos el sólido descrito dentro de la aproximación de función envolvente (EFA) (Bastard (1998)), considerando que las bandas de energías permitidas están desacopladas. La ecuación de onda para los electrones es una ecuación de Schrödinger con masa efectiva que describe el movimiento de los portadores en el potencial provocado por los átomos de impurezas ionizados localizados en una capa atómica. Como el potencial colombiano es de largo alcance, la concentración en el plano de las impurezas origina la deformación del perfil de energía potencial de la banda en la forma de un pozo de potencial para los electrones que tiene un tamaño relativamente grande con respecto a la distancia media cuadrática entre los mismos. Esto provoca que ese potencial cause el confinamiento de los electrones. La solución autoconsistente de este problema implica resolver simultáneamente las ecuaciones:

Como se observa de (2) el potencial que en el que se mueven los electrones es debido a las impurezas ionizadas (se supone que se ionizan el total de los átomos dopantes), VI (z) y a los propios electrones que existen en el gas [potencial de hartree, VX (z)]. Esto es cierto cuando la cantidad de impurezas no es especialmente alta y cuando la temperatura es baja. En este caso se puede y debe usar una descripción del potencial que incluya también los términos de canje y correlación para considerar la interacción entre los electrones asociada al principio de Pauli.1 Entonces, la aplicación de la aproximación conocida como de Thomas-Fermi resuelve este problema autoconsistente arrojando los siguientes potenciales para el δ-DQW:
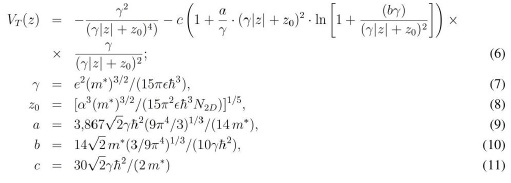
donde el primer término es el potencial de Hartree y el segundo el término de canje y correlación, con los coeficientes ajustados a partir del cálculo descrito en (Gaggero 1998).
Existen trabajos que abordan el problema electrónico en estructuras con dopamiento deltaico de forma diferente. Esto es, consideran que los átomos de impureza están distribuidos en una región de espesor no nulo, a diferencia del caso descrito aquí. Esa descripción debe, en principio, dar mejores resultados, pero depende de las demás aproximaciones que se han realizado para incorporar otros efectos (González 1996; Hai et al. 1995; Sahu 2000; Sahu 2004). Según estos trabajos, estas mejoras de la descripción del sistema no introducen modificaciones sustanciales en los resultados de los problemas que se presentan, los que ya fueron descritos en la introducción.
Si se trabaja a bajas temperaturas puede suponerse que la dispersión por impurezas es el mecanismo principal -y predominante- de interacción con los electrones. Por otra parte, cuando se trabaja a temperatura ambiente o a altas temperaturas, es necesario incluir el efecto de los fonones y su interacción con los electrones.
El perfil de potencial que se utiliza en nuestro cálculo se muestra en la figura 1. Este potencial tiene usualmente tres o más subbandas en la BC y todas ocupadas a T = 0 K. En rigor, el número de estados (subbandas) depende de la densidad de impurezas ionizadas que tenga la capa delta dopada.

Los estados obtenidos en el δ-DQW con este potencial se muestran en la figura 2. Como puede verse, para el valor de la concentración n2D empleado, aparecen solamente tres estados ligados en el pozo.
3. Cálculo de la movilidad de electrones en un pozo delta dopado
La movilidad de baja temperatura μ puede calcularse mediante (González 1994):

donde μi es la movilidad en la subbanda i-ésima y ni es la concentración de electrones en esa subbanda. La movilidad de la subbanda está relacionada con el tiempo de relajación τi a través de (González (1994)):

donde m* es la llamada masa efectiva de los electrones en la BC. En el marco de la descripción clásica del gas de electrones, la movilidad de los portadores (con masa m* y carga e) se define como la velocidad de arrastre que los mismos adquieren, por unidad de campo aplicado. Luego (Kittel 1996):

Esta es la unidad de la movilidad en el sistema de unidades CGS, que es el que se emplea más frecuentemente para reportar esta cantidad en la literatura científica.
El cálculo del tiempo de relajación puede abordarse a partir de evaluar distintas expresiones -equivalentes- determinadas bien por el marco representativo (esquema del momentum, de la energía, etc.) o por la simetría en el problema (Mitin 1999):

En todos estos casos, las  cantidades son las llamadas tasas de dispersión (en inglés, scattering rates), que establecen la probabilidad de transición de un electrón desde
cantidades son las llamadas tasas de dispersión (en inglés, scattering rates), que establecen la probabilidad de transición de un electrón desde un estado inicial
un estado inicial  hacia un estado final producto de un evento de dispersión. Además, en la expresión (19), μ representa el potencial químico del sistema estadístico.
hacia un estado final producto de un evento de dispersión. Además, en la expresión (19), μ representa el potencial químico del sistema estadístico.
Como se observa, los procesos de relajación tienen relación directa con los mecanismos de colisiones mediante los cuales el sistema tiende al equilibrio y, en este caso, se ha supuesto que el mecanismo que actúa es la interacción electrostática con los iones. Como es conocido, en el límite de muy bajas temperaturas sólo ocurrirán procesos dispersivos que modifican el espectro de energías en el entorno de la energía de Fermi.
En el cálculo de interés debemos hallar τi(E) para obtener la movilidad mediante la ecuación (13). Así, usamos la expresión (16) y consideramos que trabajaremos en el caso degenerado; para obtener el tiempo de relajación y con él la movilidad como función de la concentración de impurezas de la capa delta-dopada.
Supondremos como se hace habitualmente que tenemos un proceso de dispersión el 'astica y escribimos para la magnitud de la correspondiente tasa de dispersión la expresión proporcionada por la Regla de Oro de Fermi:

Los estados monoelectrónicos vienen dados por la expresión:

donde las Φn(z) son del tipo de las funciones mostradas en la figura 2.
Concentrémonos en el elemento de matriz que aparece en la expresión (20), esto es:
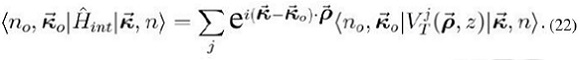
donde hemos hecho explícito que esto corresponde al potencial de una impureza colocada en el plano; pero que es necesario sumar por todas las impurezas, dado el carácter de largo alcance de la interacción de coulomb. (Esta notación sólo se empleará en esta expresión, porque no es necesaria más adelante).
El potencial correspondiente a esta interacción es el potencial apantallado de una impureza que se calcula mediante: (Fernández-Velicia 1996; Rodríguez-Coppola 2000)

En todo lo anterior hemos escrito la función dieléctrica inversa expresada en la representación de las funciones de largo y corto alcances, desarrollada en (Fernández-Velicia 1996; Rodríguez-Coppola 2000; Rodríguez-Coppola 2005). Ahí se tiene: las funciones de corto alcance (26); las funciones de largo alcance (27); los elementos de la matriz de la función inversa dieléctrica (28); la matriz dinámica relacionada con la transición (29); el coeficiente de los elementos de la matriz dinámica (30); la expresión de la matriz dinámica a T = 0 K (31) y los elementos de matriz del producto de las funciones de corto y largo alcances (32).

Para completar todos los elementos que permiten hacer el cálculo en forma detallada, debemos escribir la forma en la que queda el potencial apantallado. Usando (23) tenemos:
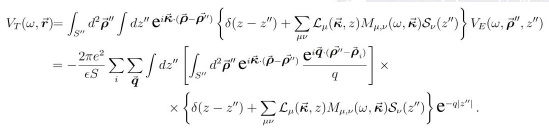
Pero realizando las integrales en forma individual se obtiene, después de sumar con la delta de Dirac:

Como puede verse en (33), calcular la función dieléctrica inversa conduce a una expresión que combina las funciones del potencial coulombiano con las funciones de onda del sistema físico bajo estudio. Este cálculo no es más complejo que otro similar que se haga, mientras que mantiene como característica distintiva el carácter tridimensional de los estados que involucra y requiere poner toda la atención en la forma en que se realizan las operaciones.
Para dar el toque final a este potencial debemos completar la suma por las impurezas y la suma por  Entonces, llegamos a:
Entonces, llegamos a:

Donde Ω(q): es la expresión entre llaves en el potencial (33). Entonces;

en tanto que, por la presencia de la se  obtiene:
obtiene:
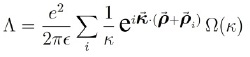
Resta por realizar la suma por todas las impurezas incluidas en el plano de dopaje (suma por i). Por ser más directo el resultado, la efectuaremos cuando calculemos el elemento matricial del potencial apantallado en el sistema. Ahora, vamos a escribir para el potencial apantallado, cuyos elementos matriciales aparecerán en el cálculo de la tasa de dispersión para hallar el tiempo de relajación, la expresión:

Antes de presentar con detalle los elementos del formalismo que anteceden al cálculo de la movilidad (y que resultan la base del mismo), es importante que describamos la fenomenología del proceso. Así, cuando se aplica un campo eléctrico a un sistema semiconductor se produce una corriente eléctrica matemáticamente dada por la expresión:

la cual se obtiene partiendo de la idea que la densidad de corriente puede escribirse como  Entonces, si suponemos que la geometría de la banda se describe en la aproximación parabólica,
Entonces, si suponemos que la geometría de la banda se describe en la aproximación parabólica,  ; la suma por
; la suma por  en (35) se realiza en la forma:
en (35) se realiza en la forma:

La dirección en que se calcula la  es la del campo eléctrico,
es la del campo eléctrico, en tanto el ángulo es el que forma con el campo. Por consiguiente
en tanto el ángulo es el que forma con el campo. Por consiguiente

Comparando la ecuación anterior con tenemos;

En las expresiones anteriores vale la pena hacer las siguientes consideraciones:
◊ El campo eléctrico tiene una dirección dada y la densidad de corriente que se calcula está en la misma dirección del campo.
◊ La única suposición realizada en el cálculo es que las subbandas del semiconductor tienen una forma parabólica. Esto hace más sencillo el problema de calcular la movilidad.
◊ La expresión para nn define la concentración areal de portadores en cada subbanda, vinculada con la cantidad de estados que existe desde su valor de energía en κ= 0 hasta el valor κfn correspondiente a cada subbanda.
◊ La expresión de la movilidad hallada juega con el tiempo de relajación de los portadores, que en este caso corresponde con la dispersión por impurezas al nivel de la energía de Fermi, pero lo que se calcula es el inverso de ese tiempo, que es para el que se tienen expresiones, por cierto nada sencillas, en dependencia de la forma en que se produce la dispersión de los portadores.
4. Cálculo estadístico de la función de distribución fuera del equilibrio
En el procedimiento anterior es necesario trabajar con un sistema que está fuera del equilibrio, por lo que la función de distribución que determina la estadística de los electrones tiene que ser calculada especialmente en estas condiciones. Para ello se emplea la aproximación lineal (Kadanoff 1999) y se considera la función de distribución real. Hacer esta consideración representa suponer que el sistema responde linealmente al campo aplicado, lo que ocurre cuando este no es muy intenso. Entonces, vamos a tener que:
* Ante un estímulo externo los portadores se mueven, y manifiestan una movilidad, en situación de no equilibrio. La distribución de portadores en el equilibrio tiene una función de distribución que llamaremos fn(0). Cuando se aplica un campo eléctrico de no mucha intensidad para mover los portadores, la función de distribución de no equilibrio cumplirá con:
Suponiendo que la función de distribución no depende del tiempo, ni de las coordenadas, entonces los cambios se producen únicamente por choques (o colisiones), luego:
pero:
Por consiguiente, se tiene:
Por otra parte, suponiendo la banda parabólica, el gradiente de la función de distribución es:
Con lo cual se escribe:
La ecuación de Boltzmann, en las condiciones enumeradas al inicio de esta sección (Kadanoff 1999), plantea que:
* Ahora, teniendo en cuenta que:
y que los índices de subbanda son mudos porque se suma sobre ellos; al sustituir en el miembro izquierdo de (39) el resultado de (38), y en el miembro derecho aquel obtenido en (20), al escribir apropiadamente los términos llegamos a:
Esta expresión se trabajará en la forma siguiente:
◊ Se hace coincidir el eje
con el campo aplicado, entonces:
, con lo cual el campo eléctrico aparece en ambos miembros de la expresión y se puede cancelar. De esta manera, el resultado no depende del campo aplicado.
◊ Para tratar el sistema se multiplica por κx, se suma por y
◊ En el miembro izquierdo de esta última relación tenemos, considerando la degeneración por el spin:
◊ Usando lo obtenido como LHS y para el otro miembro el resultado (40), escribimos:
Ahora nombramos la siguiente magnitud:
con lo que la expresión que resulta de la transformación realizada a la ecuación (39) va a quedar en la forma:
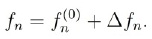





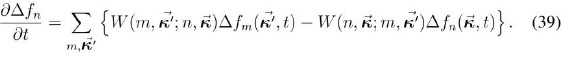

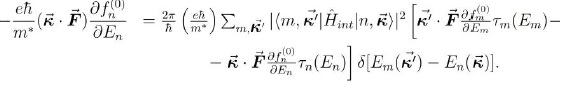






En (42) queda por terminar de evaluar el término definido por la expresión (41), a lo que dedicaremos la siguiente sección, dada su importancia y las aproximaciones que se deben hacer para calcularlo. Esa expresión es el sistema de ecuaciones algebraicas que permite calcular el tiempo de relajación de cada subbanda y con ellos la movilidad de la subbanda, para luego poder hallar la movilidad total.
5. Cálculo de las movilidades a partir de los tiempos de relajación
En este punto del análisis debemos discernir cómo calcular los elementos matriciales que aparecen en (20) -y se expresan en la forma (22)-; donde se emplea el potencial apantallado dado por (34). Además, se usan las expresiones (25) y (24) para acomodar convenientemente el resultado hallado y, en ese caso, tenemos:

donde se va a aplicar la propiedad:

y se va a introducir la función Πn,n0 :
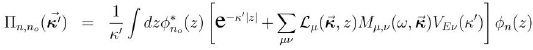
donde las diferentes funciones incluidas se definen como:
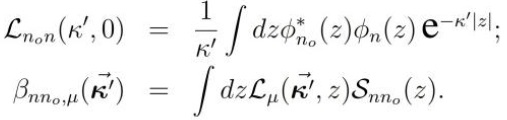
Entonces, se obtiene para Πn,n0 (') la expresión,

y esto permite escribir la correspondiente expresión para los elementos matriciales de interés en la forma:

que es no nulo sólo si ' = 0 - , por lo que va a quedar:

En este punto debemos tener en cuenta que, como el potencial es simétrico y los estados monoelectrónicos tienen paridad definida, hay elementos matriciales que se anulan y deben seguir las reglas conocidas para el cálculo de los elementos matriciales para  que ya estos autores analizamos al calcular la función dieléctrica del pozo delta-dopado (Rodríguez-Coppola 2010).
que ya estos autores analizamos al calcular la función dieléctrica del pozo delta-dopado (Rodríguez-Coppola 2010).
Ahora, con esta expresión (43) se pueden determinar los elementos matriciales buscados, (41):

pero podemos simplificar la expresión si promediamos mediante la siguiente aproximación:
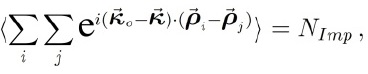
con lo cual, si usamos una de las superficies para plantear: N2D=NImp/S obtenemos:

Como ya hemos mencionado, emplearemos el esquema utilizado por M. Mora-Ramos en (Mora-Ramos 2008). Renombramos a (43) en la forma:
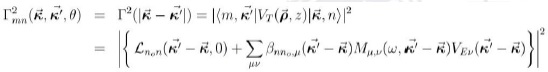
dado que este elemento depende, en realidad, de  . Así, vamos a tener para la doble suma por las variables de momentum:
. Así, vamos a tener para la doble suma por las variables de momentum:

Debemos resaltar que esta expresión para Γ2mn; constituye un coeficiente adimensional que no participa en la determinación de las unidades de los términos de interés. Por otra parte, en (41) quedan integrales por el momentum que deben completarse mediante el siguiente procedimiento: Se efectúa el cambio de coordenadas angulares de (θ, θ') a (θ, α = θ' - θ). Por comodidad, tomamos θ ∈ (0, 2π) y α ∈ (θ, π). Esos cambios gráficamente corresponden a lo mostrado en el esquema del sistema de coordenadas mostrado (Ver figura 3).
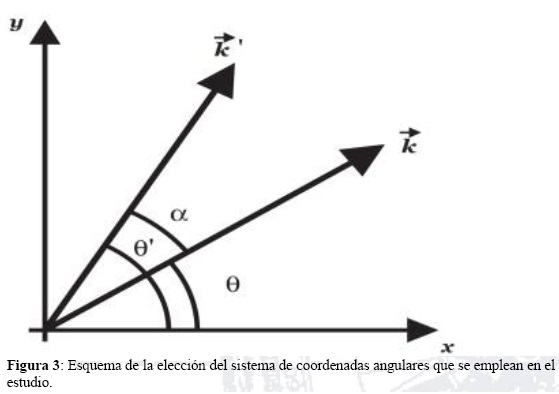
En este caso tenemos:

Con el cambio de variables angulares y considerando que:
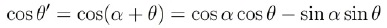
más la definición

las integrales Imn son:

Las integrales angulares pueden ejecutarse directamente. Recordando que en un proceso de dispersión elástica se |K| = |k'|cumple entonces:

Ahora, se utiliza el argumento de que a bajas temperaturas sólo los vectores de onda en una franja estrecha alrededor de la superficie de Fermi participan en el proceso de dispersión, de modo que vamos a  escribir y se tiene:
escribir y se tiene:

así que nos queda solamente por acometer la integración angular por α. Entonces tenemos:

y efectuando se llega a:
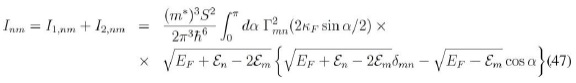
Con todo esto, el elemento matricial de interés es, usando (47):

Al pasar a la evaluación numérica, la expresión que se programará para la obtención de Kmn será:

Aquí, la cantidad Jmn(E) es un factor de energía que varía, en tanto H es una constante que depende de la concentración de impurezas del plano dopado.
Debemos significar, de forma general, que los elementos Knm son simétricos dado que representan elementos matriciales de un potencial simétrico entre estados monoelectrónicos. Esto facilita el cálculo porque simplifica las ecuaciones que hay que usar. Además, como veremos cuando escribamos explícitamente los sistemas de ecuaciones que se requieren para diferentes densidades de dopaje, las matrices que hay que usar para resolver los sistemas lineales son sencillas porque tienen muchos elementos nulos. Entonces, solamente resta por evaluar una integral angular para completar el estudio. Podemos avanzar un poco más si usamos el teorema del valor medio para el elemento matricial y escribimos:

Aquí quedan por ejecutar las integrales:

con lo cual se obtiene:

En este caso, sólo se tienen contribuciones intrasubbanda a los tiempos de relajación. Esto es, se cumple que:

Debe decirse aquí que si nos limitamos al SQL, esto es, considerar solamente la primera subbanda tenemos, de forma general, la reproducción del resultado que aparece reportado en diversos trabajos anteriores (Mora-Ramos 2008). Esto se obtiene porque ese resultado no depende de la forma concreta que tiene el elemento de matriz del potencial apantallado que se utiliza en el cálculo del inverso del tiempo de relajación. Por consiguiente, se puede decir que el cambio en la forma en que se considera el apantallamiento de la interacción coulombiana entre portadores no modifica la forma general del cálculo del tiempo de relajación, aunque sí su valor, dado que al resultado que se obtiene le falta incluir la contribución proveniente de ejecutar una integración. Esta va a proporcionar diferentes valores de acuerdo a la forma que tenga el elemento matricial considerado.
Para dar un ejemplo de la manera de actuar en el cálculo que resta, debemos considerar el problema con varias subbandas ocupadas -y, de hecho, varias subbandas en el rango de energías de interés. Veamos:
En un pozo con n2D = 1013 cm-2 en GaAs dopado con Si se tienen 6 estados del espectro discreto en la BC. Considerando sólo estos estados habrá que tener en cuenta transiciones de todos entre sí. Supongamos que sean, de hecho, seis. Entonces este sistema queda:

Usando (13) tendríamos, en nuestro caso específico:

Con esto, el algoritmo para el cálculo numérico se resume como:
- Calcular los estados electrónicos del pozo delta dopado correspondiente a una determinada concentración de impurezas en el plano. Se determinan las energías y las funciones de onda de cada estado. El rango de concentraciones de impurezas debe ser n2D = 9x1011-1013 cm -2 El valor de la concentración será uno para cada cálculo hasta el final.
- Calcular los elementos matriciales que aparecen en la función dieléctrica con el potencial del pozo y con el potencial de interacción coulombiana entre partículas
- Formalizar las expresiones (48) para hallar las integrales que aparecen, usando los elementos calculados.
- Formalizar el sistema de ecuaciones algebraicas (51) para hallar los tiempos de relajación de cada subbanda y con ellos las movilidades de cada subbanda.
- Hallar la movilidad de baja temperatura del sistema mediante la expresión (53), sumando las movilidades de cada subbanda presente en el cálculo. Se debe hacer un gráfico de la dependencia de la movilidad del sistema con la concentración de impurezas en la capa dopada. Este gráfico es el dado por otros autores (González 1994; Shi 1997).
6. Conclusiones
Después de haber expuesto el procedimiento a desarrollar para obtener la movilidad de los portadores en el sistema podemos decir lo siguiente:
- De forma natural el esquema de cálculo conduce a la obtención de una movilidad para los portadores en cada subbanda. Como se ve, este valor tiene contribuciones de todas las subbandas, aunque estas contribuciones no son visibles en forma sencilla porque realmente vienen incluidas en el apantallamiento del potencial para el cálculo de los elementos matriciales del potencial de interacción.
- Nuestro cálculo demuestra en forma precisa que las contribuciones a la movilidad de los portadores en una subbanda no transcurren a través de procesos virtuales en que el electrón salta de una subbanda a otra o al continuo y regresa a la subbanda porque estas posibles contribuciones resultan nulas, lo que simplifica el sistema algebraico que se utiliza para el estudio. Esto, además, significa que las interacciones inmediatas de los portadores de carga son solamente entre portadores de la misma subbanda y los demás portadores sólo influyen a través del apantallamiento de la interacción que se tiene en cuenta.
- Los otros esquemas de cálculo no tienen en cuenta el carácter 3D de los estados y de las magnitudes planteadas y ello no permite que puedan dar estas conclusiones sin terminar el cálculo completo.
- En nuestro cálculo no se ha tenido en cuenta lo que puede ocurrir con el continuo de energía de la banda. En rigor deben poder existir procesos en los que el electrón pase de un estado en una subbanda al continuo y de regreso a la subbanda para dar su aporte a la contribución; pero supondremos que estas son menores en valor relativo y por ello no las consideraremos.
Referencias
[1] Bastard, G. (1998). "Wave Mechanics Applied to Semiconductor Heterostructures", Ed. Les éditions de physique, Les Ulis Cedex. [ Links ]
[2] Fernández-Velicia, F.J., F. García-Moliner, and V. R. Velasco. (1996), Phys. Rev. B 53, 2034. [ Links ]
[3] Gaggero-Sager, L.M., (2002). Phys. Stat. Sol. (b), 231, 243. [ Links ]
[4] Gaggero-Sager, L.M., M.E. Mora Ramos, and D.A. Contreras Solorio. (1998). Phys. Rev. B 57, 6286. [ Links ]
[5] González, L.R., J. Krupski, and T. Szwacka. (1994). Phys. Rev. B 49, 11111. [ Links ]
[6] González, L.R., J. Krupski, and T. Szwacka. (1996). Phys. Rev. B 54, 7658. [ Links ]
[7] González, L.R., J. Krupski, M. Pietka, and T. Szwacka. (1999). Phys. Rev. B 60, 7768. [ Links ]
[8] Hai, G.Q., and N. Studart. (1995). Phys. Rev. B 52, R2245. [ Links ]
[9] Hai, G.Q., N. Studart, and F. M. Peeters. (1995). Phys. Rev. B 52, 11273. [ Links ]
[10] Ioriatti, L. (1990). Phys. Rev. B 41, 8340. [ Links ]
[11] Kadanoff, L. P. (1999). "Statistical Physics: Statics, Dynamics and Renormalization", Ed.World Scientific, Singapore. [ Links ]
[12] Kittel, Ch. (1996). "Introduction to Solid State Physics", Ed. John Wiley & Sons, New York. [ Links ]
[13] Luttinger, J. M., and W. Kohn. (1955). Phys. Rev. 97, 869. [ Links ]
[14] Mezrin, O., and A. Shik. (1991). Superlatt. Microstruct. 10, 107. [ Links ]
[15] Mitin, V. V., V. A. Kochelap and M. A. Stroscio. (1999)."Quantum heterostructures: Microelectronics and optoelectronics", Cambridge University Press. Cambridge & New York. [ Links ]
[16] Mora-Ramos, M. E. (2008). "Notas sobre el problema de la dispersión de electrones en las estructuras cuánticas", UAEM, Cuernavaca (sin publicar). [ Links ]
[17] Rodríguez-Coppola, H., V.R. Velasco, and F. García-Moliner. (2000), Physica Scripta 62, 200. [ Links ]
[18] H. Rodríguez-Coppola, F. García-Moliner, J. Tutor-Sánchez, (2005), Microelect. J. 36, 778. [ Links ]
[19] Rodríguez-Coppola, H., L.M. Gaggero-Sager, M.E. Mora-Ramos and R. Pérez-Álvarez. (2010). J. App. Phys., 108, 083717. [ Links ]
[20] Sahu, T., and J. Patnoik. (2000). J. App. Phys. 88, 2658. [ Links ]
[21] Sahu, T. (2004), J. App. Phys. 96, 5576. [ Links ]
[22] Shi, J. M., P. M. Koenrad, A. F. W. van de Stadt, F. M. Peeters, G. A. Farias, J. T. Devreese, J. H. Wolter, and Z. Wilamowski. (1997), Phys. Rev. B 55, 13093. [ Links ]
1 Esta consideración supone que se tiene en cuenta la interacción electrostática (potencial de Hartree) y la correlación del movimiento de los electrones debido al carácter indistinguible de los mismos (potencial de canje y correlación).

