











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
El síndrome Meckel-Gruber (MKS) (disencefalia esplacnoquística, OMIM #249000) es una enfermedad autosómica recesiva causada por disfunción ciliar primaria durante la embriogénesis temprana. Es caracterizado por la tríada siguiente: displasia quística renal bilateral (100%), encefalocele occipital (90%) y polidactilia postaxial (83.3%).1-3 La prevalencia estimada a nivel mundial oscila entre uno en 1,300 y uno en 140,000. Su incidencia es variable, depende de la región geográfica y del origen étnico.4,5
El MKS es una alteración genéticamente heterogénea, con al menos 13 diferentes genes conocidos. Los genes con mayor repercusión son: MKS1, MKS3, MKS4 y MKS6.6
El objetivo es reportar un caso de síndrome Meckel-Gruber diagnosticado prenatalmente, el cual presentaba los hallazgos ecográficos clásicos.
Caso clínico
Femenino de 21 años, primigesta, acude por referencia de centro de segundo nivel debido a embarazo de 19.4 semanas de gestación por ultrasonido del segundo trimestre, el cual reporta feto con alteraciones estructurales. Sin antecedentes heredofamiliares de importancia, antecedentes de patología genética-reproductiva negados y antecedente de consanguinidad negativa (Figura 1).
Se realizó estudio ecográfico de alta definición (estructural), el cual reportó lo siguiente: feto de 19 semanas, por fetometría promedio (tres semanas menor), se observó microcráneo a expensas de encefalocele occipital con gran cantidad de materia protruyendo la cavidad amniótica, derrame pericárdico de predominio derecho y probable transposición de grandes vasos. El tamaño abdominal fue excesivo a expensas de nefromegalia bilateral, los pies con talones prominentes; no se observó distensión de cámara gástrica ni vejiga. Debido a posición fetal y líquido amniótico subjetivamente disminuido no fue posible confirmar la cuenta de dedos de manos y pies. La impresión de diagnóstico desde el punto de vista ecográfico fue síndrome Meckel-Gruber (debido al encefalocele occipital y nefromegalia).
Se llevó a cabo el asesoramiento genético a los padres, explicando de forma detallada y clara los hallazgos observados en la ecografía, haciendo mención de que el síndrome tiene mal pronóstico para la vida y función del paciente (feto), por lo cual solicitan la interrupción del embarazo mediante consentimiento informado.
Los familiares autorizaron la autopsia, la cual fue realizada en nuestra unidad hospitalaria. El reporte del estudio anatomopatológico mencionó lo siguiente: feto masculino de 20 semanas aproximadamente con alteraciones de sistema nervioso central (encefalocele occipital) y alteraciones en la migración neural, displasia renal quística bilateral, sin anomalías obstructivas en vías urinarias, alteraciones hepáticas con proliferación de la placa limitante (fibrosis hepática congénita), alteraciones ductales en páncreas, polidactilia postaxial izquierda (Figura 2) que constituyen Meckel-Gruber.
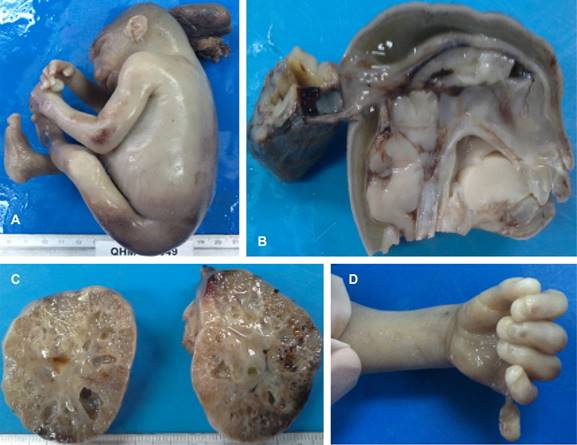
Figura 2: A) Feto con encefalocele occipital y talones prominentes. B) Encefalocele occipital (corte sagital). C) Displasia quística renal bilateral. D) Polidactilia postaxial izquierda.
De acuerdo con las características ecográficas y anatomopatológicas previamente descritas se estableció el diagnóstico definitivo de síndrome Meckel-Gruber.
Discusión
El MKS fue descrito por primera vez por Johann Friedrich Meckel en 1822 en dos hermanos que murieron de malformaciones idénticas de encefalocele occipital, riñones poliquísticos y polidactilia. George B. Gruber en 1934 informó casos familiares con características similares y acuñó el término disencefalia esplacnoquística.7),(8
El diagnóstico prenatal de MKS puede realizarse mediante ecografía del primero (11-14 SDG) o segundo trimestre (18-23 SDG). Ello obedece a que, dependiendo de las semanas de gestación, podrán ser detectables cada uno de los hallazgos correspondientes a la patología.9,10 La edad gestacional en la cual se llevó a cabo el diagnóstico en nuestro caso fue de 19 semanas, similar a lo reportado en los artículos publicados.
Los criterios diagnóstico de MKS incluyen la presencia de al menos dos de los siguientes: displasia quística renal (100%), encefalocele occipital (90%) y polidactilia postaxial (83.3%). En la mayoría de los casos el encefalocele es de localización occipital (75-80%), parietal (15-20%) y frontal (5%).11,12 En nuestro reporte se observó que los criterios de diagnóstico y la localización del encefalocele corresponden con lo previamente informado en la literatura.
Los hallazgos anatomopatológicos que se han descrito en la literatura son: displasia quística renal, encefalocele occipital, polidactilia postaxial y displasia hepática (fibrosis, infiltración linfocitaria, alteración ductal). Es importante realizar el estudio anatomopatológico correspondiente, ello permitirá corroborar y relacionar los hallazgos ecográficos, anatomopatológicos y genéticos permitiendo un diagnóstico de certeza.13,14 En nuestro caso se realizó el estudio anatomopatológico respectivo, en el cual se observaron los hallazgos característicos descritos en la literatura.
El diagnóstico diferencial del MKS se establece con trisomía 13, riñón poliquístico autosómico recesivo, Smith-Lemli-Opitz y Bardet-Biedl, debido a que comparte datos clínicos con algunas de estas patologías.15 En nuestro caso, la presencia de los hallazgos clásicos de MKS permitieron el diagnóstico del padecimiento (Tabla 1).
Tabla 1: Diagnóstico diferencial.
| Síndrome | Encefalocele occipital | Displasia quística renal | Polidactilia postaxial | Herencia | Cromosoma y/o gen | Locus |
|---|---|---|---|---|---|---|
| Meckel-Gruber | Sí | Sí | Sí | Autosómica recesiva | MKS1 | 17q22 |
| Trisomía 13 | No | Sí | Sí | Cromosómica | Cromosoma 13 | |
| Riñón poliquístico | No | Sí | No | Autosómica recesiva | PKHD1 | 6p12.3-p12.2 |
| Smith-Lemli-Opitz | No | Sí | Sí | Autosómica recesiva | DHCR7 | 11q13.4 |
| Bardet-Biedl | No | Sí | Sí | Autosómica recesiva | BBS1 | 11q13.2 |
El asesoramiento genético es un proceso importante que permite establecer el diagnóstico, pronóstico y manejo prenatal, el cual se llevó a cabo en nuestro caso. Para la posterior planificación genética-reproductiva de un embarazo se deberá ofrecer a las pacientes con mayor riesgo de recurrencia, el acceso a las pruebas de diagnóstico prenatal de acuerdo a la edad gestacional respectiva, un control prenatal estricto y las valoraciones multidisciplinarias correspondientes.

