











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
El término hipertensión portopulmonar1 (HPOP) se utiliza cuando existe hipertensión arterial pulmonar HAP (presión media en la arteria pulmonar > 25 mmHg en reposo y presión media capilar < de 15 mmHg) asociada a hipertensión portal (HPO) como una complicación de enfermedad hepática crónica,2 siempre y cuando no exista una causa alternativa de HAP como enfermedades de la colágena, enfermedad cardiaca congénita o la implicación de ciertos fármacos. Fisiopatológicamente la HAP incluye incrementos tanto de la resistencia vascular pulmonar como del gradiente transpulmonar. Se llegó al diagnóstico de HPOP en una paciente que se presentó, por tal motivo efectuamos esta comunicación.
Caso clínico
Femenino de 85 años con antecedentes de hipotiroidismo de 20 años de evolución manejada con levotiroxina 100 mg/día; niega tabaquismo. Refirió tres años de evolución insidiosa, edema de miembros inferiores, aumento de circunferencia abdominal, astenia, hiporexia, equimosis y epistaxis, manejada por facultativo con diuréticos no especificados, con disminución y recurrencia del edema de miembros inferiores. Un mes antes de su ingreso presentó tos seca acompañada de disnea de medianos esfuerzos que progresó a pequeños esfuerzos e incremento del edema de miembros inferiores abarcando tercio superior de muslos. A su ingreso a urgencias, la exploración mostró paciente disneica, ansiosa con diaforesis y presencia de áreas equimóticas en las cuatro extremidades. Peso 70 kg. TA 100/60; FC 100 lat/min FR 24 resp./min; Temp. 36 oC. Oximetría digital de 84% al aire ambiental, presencia de red venosa colateral en tórax y abdomen e ingurgitación yugular. Estertores finos en ambas bases pulmonares y ruidos cardiacos con desdoblamiento del segundo ruido en foco pulmonar y murmullo sistólico. Abdomen con red venosa colateral, aumentado de volumen a expensas de líquido intraabdominal, extremidades superiores e inferiores con edema, fóvea ++ hasta tercio superior de muslos que no permite valorar pulsos de extremidades pélvicas, llenado capilar digital de dos segundos. El electrocardiograma en urgencias mostró: ritmo sinusal con frecuencia de 100 lat/min; desviación del eje a la derecha (-150o); onda p mayor de 2.5 mV; R en V1, ST negativo y ondas T negativas en V1, V2 y V3. Los laboratoriales revelaron: Hb 12 g/dL; urea 24 mg/dL, Cr 0.5 mg/dL, colesterol 78 mg/dL, Na 132 mEq/L; tiempo de protrombina 25.5 s; INR 2.18; albúmina 2.9 g/dL y TSH 0.001 µUI/mL; anticuerpos antinucleares positivos con patrón homogéneo, anticuerpos antihepatitis y anticuerpos antimúsculo liso negativos. El gammagrama hepatoesplénico evidenció: captación hepática irregular con captación aumentada del radio trazador a nivel de retículo-endotelio extrahepático (Figura 1). La tomografía computarizada de tórax reveló: crecimiento global de cavidades cardiacas, moderado derrame pericárdico, severo derrame pleural bilateral y arteria pulmonar con calibre de 35 mm (normal = 25 mm ± 0.2) (Figura 2). La tomografía de abdomen confirmó: ascitis y dilatación de venas suprahepáticas (Figura 3). El ecocardiograma indicó presión sistólica de arteria pulmonar en 62 mmHg con presión media de 40 mmHg; la resistencia vascular pulmonar calculada en 2.7 UW; fracción de expulsión del VI de 51% y derrame pericárdico de 200 mL. Con los datos clínicos y de laboratorio se llegó a la conclusión de alta probabilidad diagnóstica de hipertensión portopulmonar en paciente con grado funcional IV de NYHA, debido a la edad y condiciones de la paciente no fue sometida a medición hemodinámica con cateterismo, ni a medición de la presión venosa hepática en cuña y libre por el alto riesgo de estos procedimientos, razón por la que se inició tratamiento sin la confirmación total de HPOP.
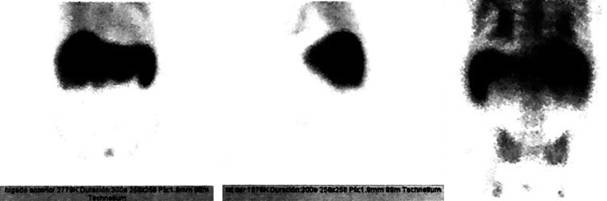
Figura 1: Gammagrafía hepatoesplénica con tecnecio marcado con sulfurocoloidal que muestra hepatomegalia a expensas de lóbulo izquierdo, concentración de material radiactivo combinado (áreas de captación normal con zonas de disminución), bazo con forma y situación normales.
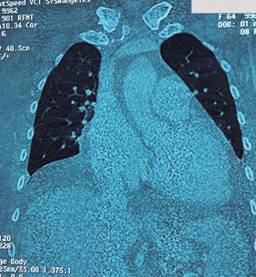
Figura 2: Tomografía computarizada de tórax y abdomen que muestra disminución del volumen pulmonar por derrame moderado, consolidación y broncograma aéreo en ambos lóbulos inferiores, además de moderado derrame pericárdico y crecimiento global de cavidades cardiacas, calcificación del botón aórtico y arteria pulmonar principal con diámetro de 35 mm en paciente femenino con hipertensión portopulmonar.
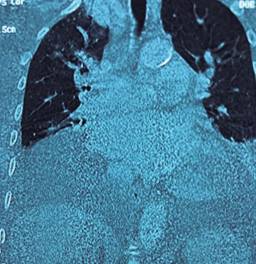
Figura 3: Tomografía computarizada de tórax y abdomen que muestra mediastino ensanchado por tejido adiposo y prominencias vasculares, hígado con dilatación de venas suprahepáticas (16 mm) y líquido de ascitis perihepático y en ambas correderas parietocólicas.
Se le manejó con: a) suspensión de tiroxina en vista de datos de hipertiroidismo, b) oxígeno 2 L/min; furosemida 40 mg IV BID; espironolactona 100 mg QD; albúmina humana al 20% 50 mL BID e inhibidores de fosfodiesterasa 25 mg TID. Su evolución mostró mejoría y fue egresada a los siete días con disminución de peso de 8 kg.
A un mes de seguimiento con ecocardiografía de control se observó: presión sistólica de arteria pulmonar en 50 mmHg y presión media de 34 mmHg, con ausencia de derrame pericárdico y fracción de expulsión de 55%; la prueba de caminata de seis minutos (no realizada a su ingreso por imposibilidad) proporcionó 210 m, correspondiendo a 52.7% del valor de referencia calculado por la ecuación de regresión de Enright,3 con frecuencia cardiaca inicial de 78 lat/min y final de 110 (81% de FC máxima permitida); FR inicial de 18 X’ y final de 24 X’; oximetría de pulso inicial de 96% y final de 92%, con disnea de tres (moderada) en escala de Borg de 0-10; FC a los 5’ 80 lat/min y FR 20X’; con grado funcional II-III de NYHA.
Discusión
La Organización Mundial de la Salud clasifica la hipertensión pulmonar en cinco grupos, el primero es denominado hipertensión pulmonar primaria, mientras que en los cuatro grupos restantes se utiliza sólo el término de hipertensión pulmonar. En el caso de la hipertensión portopulmonar (HPOP) se le clasifica como un subtipo del grupo 1 de HP primaria, a este mismo grupo 1 pertenecen otros subtipos que incluyen HP debida a fármacos y toxinas, enfermedades del tejido conectivo, infección VIH, enfermedad cardiaca congénita, esquistosomiasis, anemia hemolítica crónica, hipertensión pulmonar persistente del recién nacido, enfermedad pulmonar venoclusiva y hemangiomatosis capilar pulmonar.
En relación con la epidemiología de la HPOP, su prevalencia depende de la población estudiada, incrementándose su frecuencia en los pacientes con enfermedad hepática severa. En 17,901 autopsias de personas con cambios patológicos consistentes de HAP se encontró asociación con cirrosis en 0.7%, siendo cinco veces más de los esperado. En un estudio de 507 pacientes con HPO se detectaron 2% con HAP, la prevalencia de HPOP es más alta en pacientes con enfermedad hepática terminal que son evaluados para trasplante hepático con cifras de 3.5 a 16.1%.4
Con respecto a la etiología, para que se desarrolle la HPOP es requisito que exista previamente HAP, la enfermedad hepática sin hipertensión portal no origina HPOP; la HPO aparece en casos de cirrosis, trombosis de la vena porta y fibrosis periportal sin cirrosis. El mecanismo fisopatogénico de la HPOP5 se desconoce; la teoría más ampliamente aceptada implica la presencia de sustancias humorales como serotonina, interleucina 1, endotelina, glucagón, secretina, tromboxano B2 y péptido intestinal vasoactivo, que por el daño hepático no pueden ser metabolizadas en su totalidad, por lo que sus concentraciones plasmáticas se elevan alcanzando la circulación pulmonar a través de colaterales portosistémicas originando HPOP. Genéticamente se ha detectado un gen responsable en el brazo largo del cromosoma 2 (locus 2q33) en pacientes con HAP con carácter familiar que origina una función defectuosa en el receptor de proteína morfogénica tipo II (BMPR2),6 lo que puede contribuir al desarrollo de HPOP.
Histopatológicamente los hallazgos de HPOP y de HAP son indistinguibles, incluyendo hipertrofia de la capa media con remodelación de las paredes de la arteria pulmonar y presencia de trombosis in situ; existen dos subtipos descritos: a) la arteriopatía pulmonar plexogénica, caracterizada por hipertrofia de la media, fibrosis de la íntima y lesiones que involucran toda la pared vascular y b) la arteriopatía pulmonar trombótica, que incluye hipertrofia de la media, trombosis y fibrosis excéntrica no laminar, desconociendo si se trata de cambios evolutivos de la misma enfermedad o diferentes formas de respuesta ante la lesión.
Clínicamente la mayoría de los pacientes con HPOP tienen evidencia clínica de HAP y de HPO, pocos pacientes muestran datos clínicos de HPO solamente, siendo extraordinaria la presencia de síntomas de HAP sin evidencias de HPO.4
La HPO típicamente precede a la HAP en tiempos que varían de dos a 15 años. Los síntomas de HAP asociados a HPOP son similares a otros tipos de HAP; los descritos con más frecuencia son: disnea, síncope, dolor precordial, fatiga, hemoptisis y ortopnea; los signos más frecuentemente encontrados son: segundo ruido acentuado en el componente pulmonar, acompañado de murmullo sistólico (regurgitación tricuspídea) y edema (disfunción de ventrículo derecho), la radiografía de tórax muestra arterias pulmonares prominentes y cardiomegalia en la mayoría de los casos, en el ECG se detecta hipertrofia del ventrículo derecho, desviación de eje a la derecha y datos de bloqueo de rama derecha, sólo en 4% de los casos el ECG se reporta como normal.
El diagnóstico diferencial principal se establece con el síndrome hepatopulmonar (SHP) caracterizado por la tríada clínica: a) disfunción hepática; b) hipoxemia, determinada por una presión arterial de oxígeno menor de 70 mmHg o incremento del gradiente alveolo-arterial de oxígeno mayor de 20 mmHg y c) vasodilatación pulmonar. Tanto el SHP como la HPOP constituyen los extremos de un amplio espectro de vasculopatía pulmonar que va de la vasodilatación extrema a la vasoconstricción, ambos cuadros tienen estrecha relación patogénica con la hipertensión portal; sin embargo, sus mecanismos fisiopatológicos son exactamente opuestos y clínicamente difíciles de diferenciar sin el apoyo de estudios de laboratorio y gabinete.
En el caso que nos ocupa, debe tenerse en cuenta que la paciente mostraba hipotiroidismo (probablemente de origen autoinmune) y presencia de anticuerpos antinucleares positivos así como hepatopatía criptogénica, por lo que las posibilidades de hepatitis autoinmune y/o lúpica podrían ser factores etiológicos en el diagnóstico diferencial, observando que ambas situaciones se asocian al síndrome antifosfolípidos que puede originar HPOP por tromboembolismo crónico así como trombosis de venas suprahepáticas (en este caso se detectó dilatación de las mismas), por esta razón debe tenerse en cuenta que hubiesen sido de gran ayuda a la angiotomografía pulmonar y a la cuantificación de anticuerpos anticardiolipina y anticoagulante lúpico para descartar origen de la HPOP (en este caso los anticuerpos antihepatitis resultaron negativos, quedando la posibilidad lúpica de la hepatitis, pues había presencia de anticuerpos antinucleares).
Por otra parte, debe tenerse en cuenta que el estado hiperdinámico que mostraba la paciente asociado a las derivaciones portosistémicas, podría haber sido ocasionado y/o agravado por el uso excesivo de hormona tiroidea, ya que ésta puede llegar a causar incremento de la precarga del ventrículo derecho con hipertensión pulmonar secundaria (sin cambios en la pared vascular de las arterias pulmonares), lo anterior explicaría la buena respuesta de la paciente a la suspensión de hormona tiroidea, pues la respuesta al uso de inhibidor de fosfodiesterasa fue muy superior a la habitualmente reportada.
Para la evaluación de la HAP las pruebas diagnósticas incluyen ecocardiografía, ECG, radiografías de tórax o tomografía, pruebas de función pulmonar, polisomnografía, escaneo de ventilación perfusión o angiografía pulmonar, cuantificación de anticuerpos, prueba para VIH sida así como pruebas de funcionamiento hepático, siendo un diagnóstico por exclusión de HAP idiopática si no puede identificarse su origen.
La cateterización cardiaca derecha es necesaria para confirmar el diagnóstico de HAP y estimar su severidad. La presión en cuña venosa hepática debe medirse durante la cateterización para determinar la severidad de la hipertensión portal si se sospecha HPOP.7
Sin embargo, la correlación de la presión pulmonar sistólica estimada por ecocardiografía comparada con mediciones efectuadas por cateterización varían de 0.57 a 0.85, con un promedio de 0.74; el ecocardiograma puede sobreestimar valores con diferencia > de 10 mmHg hasta en 48% de los casos, pero continúa siendo un examen de gran utilidad que evita el riesgo de la cateterización en todos los casos, pero principalmente en pacientes de edad avanzada como en el caso presentado.8
En relación con el tratamiento de la HPOP la mayoría de las opciones de manejo se basan en los estudios de pacientes de HAP idiopática, varios ensayos aún se encuentran en proceso, de ahí que mucho del manejo se basa en la clasificación funcional del paciente.
Como medidas prioritarias en HPOP se utilizan: anticoagulantes (debido a riesgo elevado de trombosis vascular pulmonar y a enfermedad tromboembólica por estasis venosa, lento flujo sanguíneo pulmonar y crecimiento del ventrículo derecho) y diuréticos (por el incremento de volumen manifestados como ascitis y anasarca), en el caso de los primeros, diversos estudios indican que la anticoagulación puede mejorar la supervivencia; de no existir contraindicación debe buscarse un INR de 1.5 debido al incremento de riesgo de hemorragia en pacientes con enfermedad hepática crónica y el riesgo de sangrado de várices esofágicas.
Algunos avances terapéuticos utilizan diferentes mecanismos de acción, incluyendo vasodilatación, crecimiento vascular y remodelado vascular, los fármacos que se están probando en algunas series de pacientes para tratar HPOP9 son: epoprostenol, bosentán, sildenafil e iloprostol. La mayoría de las series reportadas son pequeñas en estudios observacionales, por lo que no existen datos suficientes para recomendar un fármaco específico. En general los pacientes en quienes se han aplicado estos fármacos pertenecen a la clase funcional II, III o IV de NYHA o de la OMS. Debe recordarse que los pacientes con HPOP no requieren someterse a pruebas de vaso-reactividad con vasodilatadores puros (bloqueadores de canales de calcio), ya que no existe beneficio, por el contrario pueden ocasionar hipotensión significativa por dilatación sistémica y disminución del llenado ventricular.
El epoprostenol es un potente vasodilatador con propiedades antiproliferativas y de antiagregación plaquetaria, con el uso intravenoso continuo mediante bomba de infusión se ha observado mejoría en la tolerancia al ejercicio y prolongación de la supervivencia en pacientes con HAP idiopática; en pacientes con HPOP los reportes indican mejoría en la hemodinámica, mayor tolerancia al ejercicio, incremento del gasto cardiaco, disminución de la presión arterial pulmonar y disminución de la resistencia vascular pulmonar, mejorando la clase funcional del paciente hasta por un año. Este fármaco se considera un puente hacia el trasplante hepático, los riesgos de efectos adversos son dolor mandibular, diarrea, eritema, artralgias y gasto cardiaco elevado; otros efectos indeseables más severos incluyen infección, trombosis y mal funcionamiento de la bomba de infusión, con presencia de hipertensión pulmonar por rebote, además el hiperesplenismo se ha reportado en pacientes con HPOP, por lo tanto no queda claro si el uso de epoprostenol incrementa más los riesgos en pacientes con HAP o en pacientes con HPOP.
Los fármacos como bosentán y ambrisentán son antagonistas de los receptores de endotelina A y endotelina B, siendo el segundo más selectivo para el receptor de endotelina A, los dos se administran vía oral beneficiando a los pacientes con HAP y probablemente a aquéllos con HPOP, las observaciones indican mejoría significativa en la clase funcional y la supervivencia. En un grupo de 13 pacientes tratados con ambrisentán con una media de 613 días se detectó que la presión media de la arteria pulmonar disminuyó de 50 a 41 mmHg, al igual que la resistencia vascular de 445 dinas/s/cm5 a 174 dinas/s/cm5, las pruebas de funcionamiento hepático se mantuvieron sin cambios durante 12 meses de tratamiento.
El sildenafil es un inhibidor de la fosfodiesterasa que ha demostrado beneficios en pacientes con HAP idiopática. Existen reportes de tratamiento exitosos en pacientes con HPOP a quienes se les suministró 50 mg/TID, 12 pacientes mostraron reducción en la presión arterial pulmonar media y en la resistencia vascular pulmonar a los tres meses, pero no al año, al igual que mejoría en la prueba de marcha de seis minutos, aparentemente no existen diferencias al usar sildenafil solo o combinado con prostanoide inhalado.
El iloprostol es un análogo de la prostaciclina que aporta beneficios en HAP idiopática cuando se administra por inhalación o por vía intravenosa, hay casos reportados con mejoría en pacientes con HPOP, siendo menos que los pacientes tratados con bosentán y ambrisentán.
El trasplante hepático es un tratamiento posible para los pacientes con HPOP, aunque aún no existen criterios hemodinámicos para determinar a los candidatos.10 Algunos pacientes, pero no todos, con HPOP que han recibido trasplante hepático han demostrado mejoría o normalización de la HAP, pero hay que señalar que el tratamiento médico no se ha comparado con las terapias relativamente nuevas. Cabe mencionar que la presión arterial pulmonar mayor de 60 mmHg está asociada a alto riesgo quirúrgico y resultados clínicos deficientes, mientras que las presiones arteriales elevadas moderadamente no influyen en la mortalidad después del trasplante.
Conclusión
Paciente con estado hiperdinámico con altas probabilidades de hipertensión portopulmonar en la que no se confirmó diagnóstico con pruebas específicas, con excelente respuesta al uso de inhibidores de fosfodiesterasa y en quien es probable que el factor de hipertiroidismo iatrogénico haya estado involucrado en su fisiopatogenia.

