











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La granulomatosis con poliangeítis es una enfermedad autoinmune que afecta ambos sexos, por lo general durante la quinta década de la vida.1,2 Se clasifica como localizada o sistémica y se asocia a anticuerpos anticitoplasma de neutrófilo (c-ANCA). En su forma sistémica los pacientes suelen presentar afección renal (de 38 a 70% de los casos) y respiratoria (de 75 a 93% de los casos).2 Sin embargo, también se describe la forma localizada de la enfermedad, por ejemplo en la órbita, donde los granulomas son el único signo inicial de la enfermedad (enfermedad primariamente orbitaria) o bien, durante la evolución de la misma. La afección orbitaria ocurre en 15% de los pacientes con granulomatosis con poliangeítis,1,2 con granulomas que se asocian a atrapamiento de músculos extraoculares, compresión del nervio óptico, proptosis, edema periorbitario, dacriocistitis, epífora, eritema palpebral y disminución de la agudeza visual < 20/200. El estudio de imagen más sensible para el diagnóstico es la resonancia magnética nuclear (RMN) y la biopsia del tejido que mostrarán vasculitis granulomatosa necrosante de vasos pequeños e infiltrados perivasculares.1 El régimen de tratamiento tanto en la forma localizada como sistémica se divide en la fase de inducción con ciclofosfamida y corticosteroides y la fase de mantenimiento con metotrexato o azatioprina.1
Caso clínico
Ingresó a hospitalización femenino de 42 años por disnea de medianos esfuerzos y tos productiva de un mes de evolución. Contaba con antecedente de atopia y 20 años de evolución de rinosinusitis, epífora y ojo enrojecido, diagnosticada con conjuntivitis y rinitis crónica, recibió esteroide tópico, antihistamínicos y lubricantes oculares, sin mejoría. Siete años previos a su ingreso fue valorada por otorrinolaringología y se realizó tomografía simple de nariz, senos paranasales y órbita que reveló proceso granulomatoso en mucosa nasal, senos maxilares y glándulas lacrimales, proptosis y rectificación del nervio óptico. Se valoró por oftalmología con agudeza visual bilateral 20/300 y presión intraocular 20 mmHg. Se tomó biopsia de mucosa nasal y tejido periorbitario que reportó inflamación granulomatosa. Exámenes de laboratorio, anticuerpos antinucleares, factor reumatoide, complemento y perfil tiroideo normales. EGO con eritrocituria y proteinuria mínima. Se diagnosticó con fibrosis angiocéntrica eosinofílica y desde entonces cursó con periodos de exacerbación de la sintomatología.
A su llegada al hospital presentó taquicardia sinusal, malestar general, rinorrea verdosa, disnea, tos seca, proptosis bilateral que impedía la oclusión palpebral, eritema conjuntival y epífora (Figura 1 A), tórax con hipoventilación subescapular y estertores crepitantes inspiratorios. En los exámenes de laboratorio destacaron la anemia microcítica hipocrómica de 7.6 g/dL y elevación de azoados con BUN 47 mg/dL y creatinina 3.77 mg/dL, examen general de orina con proteinuria 150 μg/dL y hematuria 150 eritrocitos/μL. Velocidad eritrosedimentación 26 mm/s, proteína C reactiva 3.8 mg/dL, procalcitonina 0.15 ng/mL. Tomografía de tórax con consolidación en lóbulo medio derecho, imágenes en vidrio deslustrado, distorsión fibrótica de la arquitectura e imágenes nodulares sugestivas de granulomas. Prueba rápida de influenza positiva para influenza tipo A. Se diagnosticó con neumonía adquirida en la comunidad, influenza tipo A y lesión renal. Inició terapia con cefepime 1 g IV cada 24 horas y oseltamivir 75 mg cada 12 horas. Tres días después mostró mejoría del cuadro respiratorio, pero deterioro en la función renal (creatinina 6.45 mg/dL, tasa de filtración glomerular 6 mL/min) se recibió serología p-ANCA y c-ANCA negativa. La ultrasonografía renal no reveló alteraciones, se tomó biopsia renal con hallazgo de proliferación extracapilar glomerular, esclerosis global, necrosis fibrinoide segmentaria, nefritis tubulointersticial, daño crónico intersticial, cilindros, atrofia tubular, inmunofluorescencia negativa para IgG, IgM, IgA, C1, C3 (Figura 2). Se concluyó una glomerulonefritis necrosante pauci-inmune, fibrosis 60%, compatible con granulomatosis con poliangeítis. Inició inducción a la remisión con metilprednisolona 1 mg/kg IV y ciclofosfamida 15 mg/kg IV con excelente respuesta, remisión de la sintomatología y exoftalmos (Figura 1 B), con duplicación de la tasa de filtración glomerular y disminución de la creatinina sérica a 3 g/dL. Se dio de alta para seguimiento en el servicio de nefrología. Al cierre de esta publicación se reporta con mejoría sustancial.
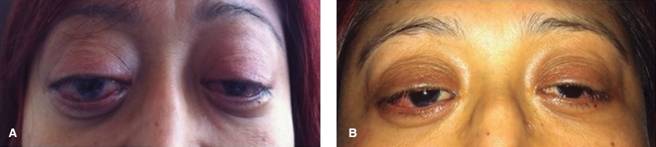
Figura 1: (A) Aspecto de la paciente el día de su ingreso. (B) Aspecto tres días después de iniciar el tratamiento inmunosupresor.
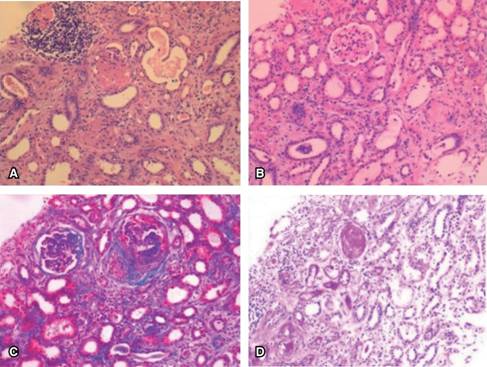
Figura 2: (A) Biopsia renal, tinción hematoxilina y eosina. Glomérulos con proliferación extracapilar y lesión necrosante. (B) Esclerosis global glomerular. (C) Tinción Masson. Nefritis tubulointersticial con fibrosis, daño crónico intersticial. (D) Tinción PAS. Necrosis fibrinoide segmentaria y atrofia tubular.
Discusión
Se presentó el caso de una paciente con granulomatosis con poliangeítis primariamente orbitaria cuyo diagnóstico se estableció 20 años después de su aparición. Inicialmente la sintomatología ocular y nasal localizadas, en ausencia de afección sistémica y pulmonar, se consideró erróneamente como fibrosis angiocéntrica eosinófila; no obstante, desde entonces la paciente ya contaba con tres de los cuatro criterios según el Colegio Americano de Reumatología (ACR 1990) para el diagnóstico de granulomatosis con poliangeítis: inflamación nasal, microhematuria e infiltrado granulomatoso en las biopsias. Adicionalmente, en el último internamiento de la paciente se agudizaron la enfermedad ocular, los granulomas evidentes, los estudios de imagenología pulmonar y la glomerulonefritis pauci-inmune, todos característicos de la enfermedad.3 Si bien la fibrosis angiocéntrica eosinófila es una entidad inflamatoria crónica y benigna de la nariz que se asocia, como en el caso de nuestra paciente, a atopia, trauma o cirugía,4,5 generalmente ésta aparece entre los 50 y 60 años de edad.6,7 Esta patología, al igual que la granulomatosis con poliangeítis, se manifiesta con obstrucción nasal, sinusitis, epistaxis y excepcionalmente lesiona la órbita5,7-10 causando proptosis, edema periorbitario, diplopía y epífora;7 sin embargo, el patrón histológico en las lesiones es un infiltrado perivascular de predominio eosinófilo y fibrosis en patrón “capas de cebolla”,4,9 lo cual no se observó en nuestra paciente. De igual manera, en la fibrosis angiocéntrica no existe afección sistémica, granulomas o vasculitis necrosante4,6-9 como se evidenció en la biopsia renal del caso aquí presentado. En cuanto al tratamiento, se sabe que en la fibrosis angiocéntrica los inmunosupresores no proveen mejoría4,6,9 como se demostró en este caso.
Es importante resaltar que en este caso, la lenta evolución de la enfermedad hacia la pérdida visual, lesión pulmonar y renal progresivas, a pesar de no haber recibido inmunosupresores, se explica parcialmente por la falta de seropositividad serológica que confiere un mejor pronóstico en los pacientes y que incluso en ausencia de anticuerpos fue posible confirmar el diagnóstico, ya que los c-ANCA tienen una sensibilidad de sólo 66%1 y falsos negativos hasta de 25 a 48% de los casos.2
Conclusiones
La granulomatosis con poliangeítis primariamente orbitaria es una patología rara cuyo diagnóstico clínico requiere criterios clínicos, serológicos, histopatológicos y de imagen. Es importante establecer el diagnóstico certero y oportuno de las lesiones oculares cuando se presentan como único dato inicial de la enfermedad a fin de prevenir las complicaciones irreversibles.

