











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Organic chemists, inspired by Nature creativity, have created a variety of methodologies and strategies for the synthesis of chemical molecules. Huisgen generalized 1,3-dipolar cycloaddition reaction in the 1960s [1, 2]. which is among the most potent and flexible approaches for the highly selective synthesis of five-membered heterocyclic compounds. 32CA reactions are bimolecular in nature and include the 1,3-addition of an alkene derivative to a three-atom component (TAC), yielding five-membered heterocycles. which are adaptable intermediates in the production of natural products as well as biologically and medicinally active chemicals [3]. The 32CA reaction of nitrones with alkenes is the most often used technique for the synthesis of isoxazolidines, which are readily converted to 1,3-aminoalcohols owing to the existence of a labile N-O link, [4] aminoacids [5] and alkaloids as intermediates in their production [6]. The measurement of variations in electron density along the reaction route with the aim of explaining chemical reactivity. The advantage of this approach is that electron density is both a local function specified within the precise many-body theory and an empirically accessible scalar field, which allows a good description of bonding changes to characterize a reaction process [7]. Domingo introduced the Molecular Electron Density Theory (MEDT) [8]. In this area in 2016, establishing that the feasibility of changes in electron density along a reaction route is accountable for reactivity in Organic Chemistry [9,10]. Following Woodward and Hoffmann's classification of pericyclic reactions in 1969, in which "all first order modifications in bonding interactions occur simultaneously on a closed curve," [11]. The 32-step CA reactions were classed as pericyclic reactions based on their electrical properties being similar to those of Diels-Alder reactions, namely the presence of [4 + 2] p electrons [12,14].
However, several recent MEDT experiments of 32CA reactions have shown that the bonding changes occur sequentially, rather than "concentrated on a closed curve," ruling out the pericyclic process [9] Recent MEDT studies devoted to elucidating the reactivity of three-atom-components (TACs) involved in 32CA reactions with alkene have enabled the establishment of a useful classification system for these non-polar cycloaddition reactions into pseudodiradical-type (pdr-type), pseudoradical-type (pmr-type), carbenoid-type (cb-type), and zwitterionic-type (zw-type), based on the TAC's electrical structure [15]. Recent MEDT investigations of nitrone 32CA reactions have shown that these reactions occur through an uncoordinated one-step process in which the two new single bonds are generated in an asynchronous manner [9].
The MEDT study is presented in five sect, 3.1 to 3.5, in this research work.
(I) in Sect. 3-1, the electron localisation function (ELF) at the ground state structures of reagents N-methyl-C-3-bromophenyl- nitrone 1 and dimethyl maleate 2 is topologically analyzed so that they can be represented in terms of their electronic structure and subsequently evaluated for their reactivity in 32CA reactions [16-20]. (II) In Sect. 3.2, reactivity indices described within the conceptual density functional theory (CDFT) are examined in order to understand the polarity of the 32CA reactions [21,22]. (III) In Sect. 3.3., the potential energy surfaces (PES) along the possible stereoisomeric channels of the 32CA reactions are investigated in predicting the energy profiles, and the global electron density transfer (GEDT) at the transition states (TSs) is determined [8,23,24]. (IV) In Sect. 3.4, analyzes the ELF of the discovered TSs. (V) In Sect. 3.5, the combination of ELF and Thom's disaster, as well as Krokidis' proposed bonding evolution theory (BET), is used to structure the process for electron density changes along stereoisomeric routes [1,9,25-27].
Computational methods
The Berny analytical gradient optimisation approach was used at the MPWB95/6-311++G (d,p) level for the optimization of the stationary points along the potential energy surface of the 32CA reactions [28,29]. The use of MPWB95 functional in the analysis of 32CA reaction has been shown to be a reliable and accurate approach [20,21,30-34]. The optimized TSs' frequency computations revealed the existence of a single imaginary frequency, while the disappearance of imaginary frequencies was shown to be the local minimum. Intrinsic Reaction Coordinate (IRC) computations were performed using the Gonzales-Schlegal integration technique to validate the least energy reaction route connecting the reactants and products through the identified TSs. [35-37].
The effects of the solvent on toluene were investigated using a polarizable continuum model (PCM) and the self-consistent reaction field (SCRF) approach [38-42]. The CDFT indices are determined using the equation discussed in Reference 44 [43, 44]. The global electron density transfer (GEDT) [45] was used to compute the global electron density at the TSs of each reacting framework using natural population analysis (NPA): {45-47]:
Where q signifies atomic charges, the total of charges on all atoms in the studied framework indicates the GEDT, and the positive sign of GEDT indicates global electron density flux from that framework to another [45].
Multiwfn software [36] is used to perform the topological analysis of the ELF at the reagents, TSs, and IRC sites, as well as the computation of the surfaces are shown using UCSF Chimera software [38]. All computations were performed using the Gaussian 16 software [38].
Results and discussion
ELF topological analysis N-methyl-C-3-bromophenyl- nitrone 1 and dimethyl maleate 2
In 1990, Becke and Edgecombe developed the ELF concept, which was shown in 1994 by Silvi and Savin to establish a quantifiable relationship between electron density distribution and molecule chemical structure [48,49]. A reasonable connection has been established between the electronic structure of three atom components (TACs) and their reactivity in 32CA reactions using the MEDT framework [8,9,50]. In this article, the ELF of the ground state structures of the reagents 1 and 2 are investigated to characterize their electronic structures and reactivity in 32CA reactions. Table 1 lists N-methyl-C-3-bromophenyl-nitrone 1 and dimethyl maleate 2 have significant ELF valence basin populations, whereas the domains of ELF localization are shown in Fig. 1. ELF valence populations are used to determine the Lewis structures of reagents. and are given in Fig. 2. The ELF N-methyl-C-3-bromophenyl-nitrone 1 shows the presence of two monosynaptic basins, V(O1) and V'(O1) integrate 5.8 e, a disynaptic basin V(C3, N2) integrates 3.76 e, and a disynaptic basin V(N2,O1) integrates 1.98 e, which corresponds to the non-bonding electron density on O1 oxygen, the underpopulated C3-N2 double bond, and the underpopulated N2-O1 single bond, respectively. The ELF of dimethyl maleate 2 demonstrates a single disynaptic basin for the C4-C5 bonding region with a total integrating population of 3.36 e, which is related to the underpopulated C4-C5 double bond.
Table 1 MPWB95/6-311++G(d,p) calculated most significant ELF valence basin populations at 1 and 2. ELF valence basin populations are given in average number of electrons, e.
| 1 | 2 | |
|---|---|---|
| V(O1) | 2.92 | - |
| V'(O1) | 2.88 | - |
| V (N2, O1) | 1.98 | - |
| V (N2, C3) | 1.88 | - |
| V'(N2, C3) | 1.88 | - |
| V (C4, C5) | - | 1.68 |
| V'(C4, C5) | - | 1.68 |
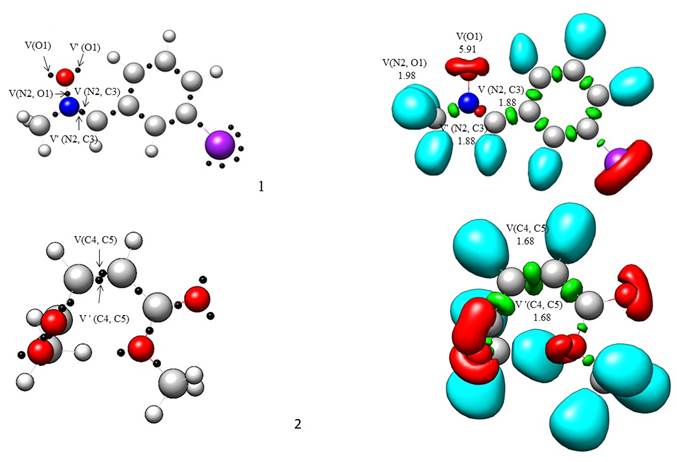
Fig. 1 MPWB95/6-311++G(d,p) ELF localisation domains represented at an isosurface value of ELF = 0.82 of N-methyl-C-3-bromophenyl- nitrone 1 and dimethyl maleate 2. Green coloured ones are the disynaptic basins, blue colour represents the protonated basins, red colour is used to represent the monosynaptic basins. The attractor positions are represented as black spheres.
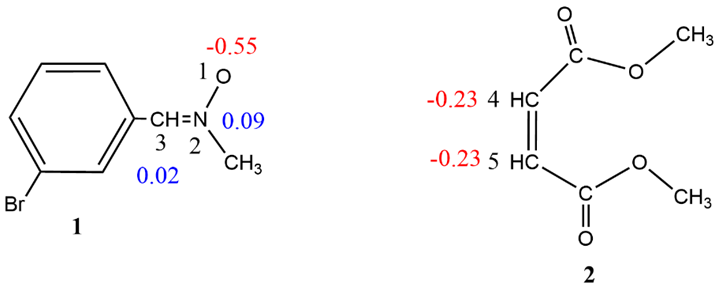
Following the establishment of the reagents' bonding patterns, the atomic charge distributions of N-methyl-C-3-bromophenyl-nitrone 1 and dimethyl maleate 2 were determined by NPA. (Fig. 2) [46, 47].The N-methyl-C-3-bromophenyl-nitrone 1 oxygen atom is negatively charged (-0.5558 e). C3 carbon shows charge of 0.0213e, while N2 nitrogen is positively charged (0.0971e). This indicates charge separation in N-methyl-C-3-bromophenyl-nitrone 1, but it differs from the charges predicted by the Lewis' bonding model. Although N-methyl-C-3-bromophenyl-nitrone 1 is classed as a zwitterionic TAC based on ELF analysis, this terminology does not refer to the nitrones' dipolar electronic structure. Rather than that, it denotes the unique bonding pattern (in the absence of charges) of Huisgen's resonance Lewis structure for 1,3-dipoles.[51]. In dimethyl maleate 2, carbon C4 and C5 shows negative charge values of -0.23043 and -0.23043, respectively.
Analysis of the CDFT indices N-methyl-C-3-bromophenyl- nitrone 1 and dimethyl maleate 2
When CDFT [44, 52] reactivity indices are analyzed, they may be used to get a first understanding of the direction of electronic flow between the reagents. At the B3LYP/6-31G(d) level of theory, the standard reactivity scales are defined, and the CDFT indices, namely the electronic chemical potential, chemical hardness, electrophilicity, and nucleophilicity N indices in eV of Accepted Manuscript 6 ACHARJEE et al. the reagents N-methyl-C-3-bromophenyl-nitrone 1 and dimethyl maleate 2, were computed at the MPWB95/6-311++G(d,p) computational level. The electronic chemical potential μ of N-methyl-C-3-bromophenyl- nitrone 1 (μ = -4.06 eV) is higher than that of dimethyl maleate 2 (μ = -5.09 eV), indicating the electronic flow along with the 32CA reaction from N-methyl-C-3-bromophenyl- nitrone 1 to dimethyl maleate 2. N-methyl-C-3-bromophenyl- nitrone 1 (N = 2.43 eV) is considered to be a moderate nucleophile, but dimethyl maleate 2 (N = 0.41 eV) is considered to be a weak nucleophile.
Both N-methyl-C-3-bromophenyl- nitrone 1 (ω = 1.38 eV) and dimethyl maleate 2 (ω = 1.62 eV) are classified as the strong electrophile. As a result, along these zw-type 32CA reactions, dimethyl maleate 2 will behave as electrophiles, whereas N-methyl-C-3-bromophenyl-nitrone 1 will behave as a nucleophile, in accordance with their electronic chemical potentials.
Table 2 B3LYP/6-31G(d) CDFT indices N-methyl-C-3-bromophenyl- nitrone 1 and dimethyl maleate 2. μ, η, ω and N represent electronic chemical potential, chemical hardness, electrophilicity and nucleophilicity indices, respectively, and are expressed in eV.
| μ | η | ω | N | |
| 1 | -4.06 | 5.98 | 1.38 | 2.43 |
| 2 | -5.09 | 7.97 | 1.62 | 0.41 |
Analysis of the energy profile associated with the 32CA reactions of N-methyl-C-3-bromophenyl- nitrone 1 and dimethyl maleate 2
The 32CA reaction of N-methyl-C-3-bromophenyl- nitrone 1 and dimethyl maleate 2 can take place along two stereoisomeric paths, namely endo and exo, associated respectively with the attack of nitrone oxygen to the C4 and C5 carbon of dimethyl maleate 2 (Scheme 1). The TSs TS1 and TS2 and the products 1 and 2. Some appealing conclusions could be derived from the energy profile study (See Table 3).
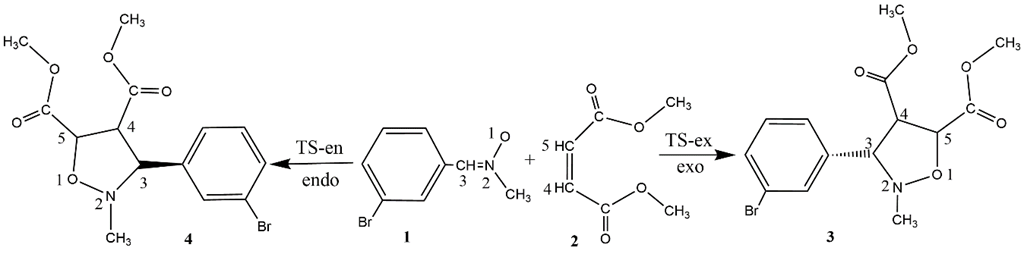
Scheme 1 Studied stereoisomeric paths for the 32 A reactions of N-methyl-C-3-bromophenyl-nitrone 1 with dimethyl maleate 2.
Table 3 MPWB95/6-311++G(d,p) relative energies (ΔE), enthalpies (ΔH), Gibbs free energies (ΔG) and GEDT (average electron population, computed as the difference between the total electronic populations of the two reacting counterparts in the transition state) of TSs and products for the 32CA reactions of N-methyl-C-3-bromophenyl-nitrone 1 with dimethyl maleate 2.
| TS | solvent | ΔE / kJmol-1 | ΔH / kJmol-1 | ΔG/ kJmol-1 | GEDT | Product | ΔE / kJmol-1 | ΔH / kJmol-1 | ΔG / kJmol-1 |
| TS-ex | Gas phase | 40.85 | 38.37 | 101.62 | 0.14 | 3 | -128.58 | -131.97 | -69.95 |
| TS-en | Gas phase | 35.47 | 34.04 | 91.99 | 0.12 | 4 | -120.76 | -123.86 | -61.13 |
| TS-ex | Toluene | 50.38 | 47.71 | 111.85 | 0.15 | 3 | -115.73 | -119.43 | -55.82 |
| TS-en | Toluene | 46.16 | 44.51 | 102.78 | 0.13 | 4 | -107.74 | -111.11 | -47.40 |
| TS-ex | Benzene | 50.01 | 47.34 | 111.44 | 0.15 | 3 | -116.25 | -119.95 | -56.37 |
| TS-en | Benzene | 45.73 | 44.07 | 102.34 | 0.13 | 4 | -108.27 | -111.64 | -47.96 |
| TS-ex | THF | 56.54 | 53.85 | 118.49 | 0.17 | 3 | -106.87 | -110.69 | -46.37 |
| TS-en | THF | 53.67 | 51.98 | 110.26 | 0.14 | 4 | -98.48 | -101.92 | -37.65 |
| TS-ex | DCM | 57.05 | 54.37 | 119.07 | 0.17 | 3 | -106.07 | -109.90 | -45.50 |
| TS-en | DCM | 54.35 | 52.67 | 110.98 | 0.14 | 4 | -97.63 | -101.07 | -36.72 |
(i) The 32CA reaction of N-methyl-C-3-bromophenyl-nitrone 1 and dimethyl maleate 2 This shows that the reaction has negative free energy ranging from -36.72 to -69.95 kJ mol-1, suggesting kinetic control and hence irreversibility.
(ii) The ΔE of TS-ex is 5.38, 4.22, 4.28, 2.87, and 2.7 kJ mol-1 higher than that of TS-en in the gas phase, toluene, benzene, THF, and DCM, respectively, suggesting exclusive endo selectivity in complete agreement with the experimental finding [53].
(iii) TS-ex has an activation enthalpy of 38.37 kJ mol-1 in the gas phase, which increases to 54.37 kJ mol-1 in DCM, 53.85 kJ mol-1 in THF, 47.34 kJ mol-1 in benzene, and 47.71 kJ mol-1 in toluene, suggesting an increase of 16 kJ mol-1 from the gas phase to DCM, thus indicating the energetically feasible reaction in low polar solvents.
The GEDT at the TSs were calculated to assess the polar character and are listed in Table 3. The GEDT values at the TSs, between 0.12 and 0.17 e, suggest that this 32CA reaction has some polar character, being classified of forwards electron density flux (FEDF) [54].
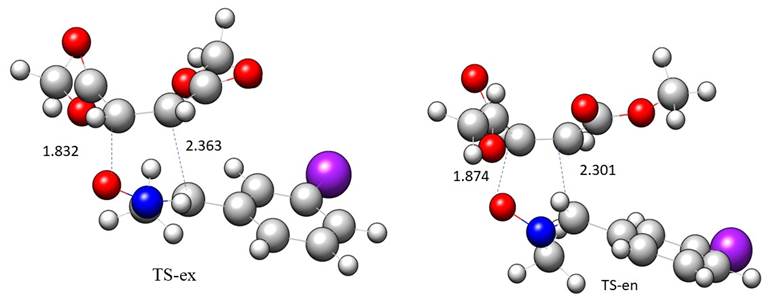
Fig. 3 shows the gas phase geometries of TS-ex and TS-en. At TS-ex, the distance between C3 and C4 interacting centers is 0.531 Å more than the distance between the C5 and O1 interacting centers. While in TS-en., the distance between the C3 and C4 interacting centers is 0.427 Å times greater than the distance between the C5 and O1 interacting centers, showing that TS-ex has a higher degree of asynchronicity than TS-en. The inclusion of the solvents has no effect on the distance between the C3 and C4 interacting centers, which is 2.363 Å in TS-ex and 2.301 Å in TS-en, and also the distance between the C5 and O1 interacting centers, which is 1.832 Å in TS-ex and 1.874 Å in TS-en.
Topological analysis of the ELF at the TSs
The ELF topological study of TS-ex and TS-en, as well as their comparison, were performed. At TS-ex and TS-en, the populations of the most important valence basins are shown in Table 4, and the ELF localization domains are depicted in Fig. 4, respectively. The ELF of the two TSs reveals the existence of two monosynaptic basins, V(O1) and V'(O1), which correspond to the O1 oxygen's non-bonding electron density. It normally occurs in N-methyl-C-3-bromophenyl-nitrone 1 with dimethyl maleate 2 (see Tables 1 and 4).
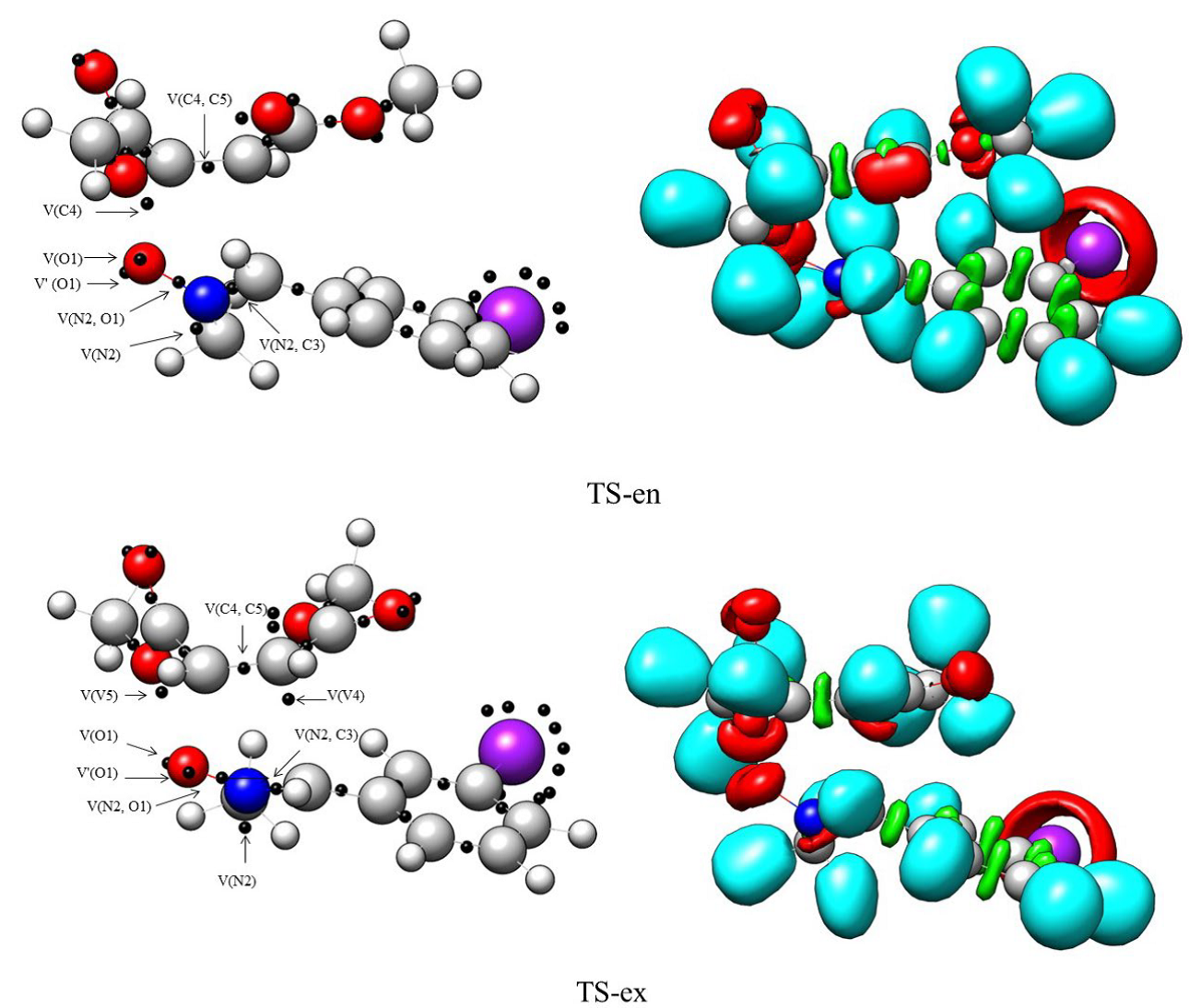
Fig. 4 MPWB95/6-311++G(d,p) ELF localization domains and the basin attractor positions of gas phase TSs TS-ex and TS-en. Protonated basins are shown in blue, monosynaptic basins in red, disynaptic basins in green and the attractor positions in magenta colour (Iso value = 0.83).
Table 4 MPWB95/6-311++G(d,p) calculated most significant ELF valence basin populations at the TSs.
| TS-ex | TS-en | |
|---|---|---|
| V(O1) | 2.85 | 2.89 |
| V'(O1) | 2.85 | 2.88 |
| V(N2) | 1.02 | 1.14 |
| V (N2, O1) | 1.31 | 1.27 |
| V (N2, C3) | 2.98 | 2.91 |
| V (C4, C5) | 2.92 | 3.13 |
| V(C4) | 0.27 |
The electron density in the N2-C3 bonding region decreases from 3.76 e in Table 1 to 2.98 e and 2.91 e in TS1-ex and TS1-en, respectively. A disynaptic basin, V(C4, C5), coupled with a C4-C5 bonding region, is identified in the dimethyl maleate 2 framework, integrating a total population of 2.92 e (TS1-ex) and 3.13 (TS1-en). The ELF of transition states reveals the existence of a single monosynaptic V(C4) basin with an integration of 0.27 e for TS-ex. as well as the V(N2) monosynaptic basin integrating 1.02 e and 1.14 e associated with the non-bonding electron density at N2 nitrogen at TS-ex and TS-en, respectively, disynaptic basins V(O1, C5) and V(C3, C4) have not yet emerged, which suggests that the formation of O1-C5 and C3-C4 single bonds has not yet begun. TS-ex and TS-en.
Mechanistic implications along the stereoisomeric channels of 32CA reaction of N-methyl-C-3-bromophenyl-nitrone 1 with dimethyl maleate 2 from bonding evolution theory (BET) study
Using the combination of the ELF [48,49] and the Thom's Catastrophe theory [55], Krokoidis [56] proposes the BET, a plausible mechanism for a chemical reaction. The BET of the energetically viable ortho route is investigated in this paper at the MPWB95/6-311++G(d,p) level of theory, which splits the reaction path into six topological phases. Table 5 shows the most important ELF valence basin populations at the beginning of each phase (P1-I - P6-I) and product 3, with a simplified depiction of the hypothesized mechanism in Scheme 2. Phase I begins at P1-I (d(O1-C5) = 2.546 Å and d(C3-C4) = 2.831 Å) and shows the existence of V(O1) monosynaptic basins that integrate a total population of 5.81 e associated with the O1 oxygen non-bonding electron density. The disynaptic basins V(N2, O1) and V(C3, N2) integrate at 1.49 e and 3.66 e, respectively, in association with the N2-O1 single bond and the C3-N2 double bond. Additionally, the ELF of P1-I reveals the existence of two V(C4, C5) disynaptic basins integrating 3.32 e in the C4-C5 double bond. Thus, the ELF of P1-I is similar to that of the separated reagents N-methyl-C-3-bromophenyl-nitrone 1 and dimethyl maleate 2 (Fig. 1) and exhibits a GEDT of 0.05 e. Phase II begins at P2-I (d(O1-C5) = 2.056 Å and d(C3-C4) = 2.512 Å). Phase III begins at P3-I (d(O1-C5) = 1.832 Å and d(C3-C4) = 2.363 Å), It is distinguished by the existence of a V(N2) monosynaptic basin integrating 1.01 e associated with nonbonding electron density at N2 nitrogen, which obtains the electron density mostly from the C3-N2 bonding region. It is worth noting that the V(C3, N2) disynaptic basin depopulates from 3.84 e in P2-I to 2.98 e in P3-I and the existence of the V(C4) monosynaptic basin integrating 0.27 e is associated with the pseudoradical centre at C4, which gets electron density from the C4-C5 bonding region. Note that the C4-C5 bonding region is depopulated from 3.2 e in P2-I to 2.92 e in P3-I. TS-ex is a constituent of this phase with a GEDT of 0.14, indicating the 32CA reaction is polar. Phase IV begins at P4-I (d(O1-C5) = 1.670 Å and d(C3-C4) = 2.234 Å) and is distinguished by the existence of a V(C3) monosynaptic basin integrating 0.26 e associated with the pseudoradical centre at C3, which is formed by deriving electron density from the C3-N2 bonding region. note that the C3-N2 bonding region has been depopulated from 2.98 e in P3-Ito 2.43 e in P4-I there is also the presence of a V(C5) monosynaptic basin integrating 0.64 e associated with the pseudoradical centre at C5, which is formed by deriving electron density from the C4-C5 bonding region. It is noted that the C4-C5 bonding region has been depopulated from 2.92 e in P3-I to 2.37 e in P4-I. Phase V begins at P5-I (d(O1-C5) = 1.541 Å and d(C3-C4) = 2.073 Å) and is distinguished by the formation of the V(O1, C5) disynaptic basin, which integrates 1.02 e associated with the O1-C5 single bond. note that the pseudoradical centers at O1 and C5 combine to create the O1-C5 single bond at a distance of 1.541 Å as a result, the V(C5) monosynaptic basins are not apparent in this phase. Phase VI begins at P6-I (d(O1-C5) = 1.431 Å and d(C3-C4) = 1.692 Å) and is distinguished by the formation of a V(C3, C4) disynaptic basin that integrates 1.68 e associated with the C3-C4 single bond. note that the pseudoradical centers at C3 and C4 pair to form the C3-C4 single bond at a distance of 1.692, the V(C3) and V(C4) monosynaptic basins are absent in this phase. suggesting a significant degree of asynchronicity in the process of bond creation. This is consistent with the fact that the C3-C4 bond distance is greater than the O1-C5 bond distance.
Table 5 ELF valence basin populations of the IRC structures P1-P6 defining the six phases, characterising the molecular mechanism of the (TS-ex) reaction path of 32CA reaction of N-methyl-C-3-bromophenyl-nitrone 1 with dimethyl maleate 2. The distances of the forming bond distances are given in angstrom units, Å.
| Phases | I | II | III | IV | V | VI | |
|---|---|---|---|---|---|---|---|
| Structures | P1-I | P2-I | P3-I TS-ex | P4-I | P5-I | P6-I | 3 |
| d(C3-C4) | 2.831 | 2.512 | 2.363 | 2.234 | 2.073 | 1.692 | 1.588 |
| d(O1-C5) | 2.546 | 2.056 | 1.832 | 1.670 | 1.541 | 1.431 | 1.415 |
| GEDT | 0.05 | 0.13 | 0.14 | 0.09 | 0.02 | 0.20 | 0.22 |
| V(O1) | 2.99 | 2.91 | 2.85 | 2.74 | 2.64 | 2.55 | 2.52 |
| V’(O1) | 2.82 | 2.86 | 2.85 | 2.60 | 2.42 | 2.38 | 2.32 |
| V (C3, N2) | 3.66 | 3.84 | 2.98 | 2.43 | 2.17 | 1.84 | 1.83 |
| V (N2, O1) | 1.49 | 1.41 | 1.31 | 1.18 | 1.12 | 1.02 | 1.03 |
| V(N2) | 1.01 | 1.53 | 1.77 | 2.18 | 2.21 | ||
| V (C4, C5) | 1.66 | 1.60 | 1.46 | 2.37 | 2.15 | 1.95 | 1.94 |
| V’ (C4, C5) | 1.66 | 1.60 | 1.46 | ||||
| V(C3) | 0.26 | ||||||
| V(C4) | 0.27 | 0.53 | 1.18 | ||||
| V(C5) | 0.64 | ||||||
| V (O1, C5) | 1.02 | 1.26 | 1.27 | ||||
| V (C3, C4) | 1.68 | 1.77 |
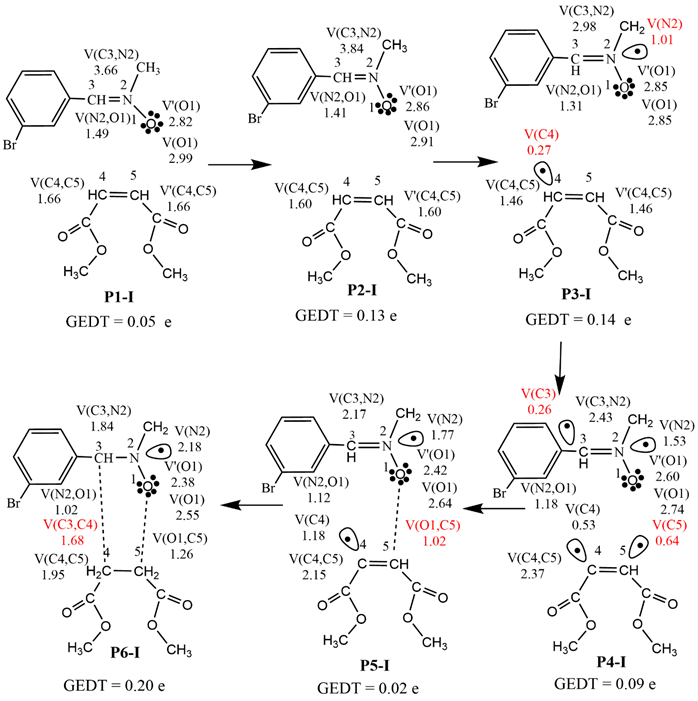
Scheme 2 Sequential bonding changes and most significant valence basin populations in average number of electrons e at the representative IRC points along the exo reaction path of the 32 CA reaction of N-methyl-C-3-bromophenyl-nitrone 1 with dimethyl maleate 2.
Scheme 3 shows the ELF basin populations at the reacting centers for the 32CA reaction of N-methyl-C-3-bromophenyl-nitrone 1 with dimethyl maleate 2 along the endo stereoisomeric reaction path, as well as the sequential bonding modifications. The identification of catastrophes using ELF basin analysis enables the characterisation of six topological phases, I, II, III, IV, V, and VI, as indicated by the beginning points P1-II, P2-II, P3-II, P4-II, P5-II, and P6-II, respectively (Scheme 3). The ELF topology of the starting point P1-II, like the individual reagents, has the same bonding pattern as the individual reagents (see Table 1). (d(C3-C4) = 2.833 Å, d(O1-C5) = 2.511 Å is derived from the C3-N2 bonding region at P2-II, showing a depopulation from 3.75 e at P1-II to 3.91 e at P2-II. phase P3-II is characterized by the formation of monosynaptic basins V(N2) integrating 1.14 e, while phase III is characterized by the emergence of pseudoradical centers at N2. noted that the C4-C5 bonding region has been depopulated from 3.14 e at P2-II to 3.12 e at P3-II, while the C3-N2 bonding region has been depopulated from 2.91 e at P3-II to 2.32 e at P4-II.
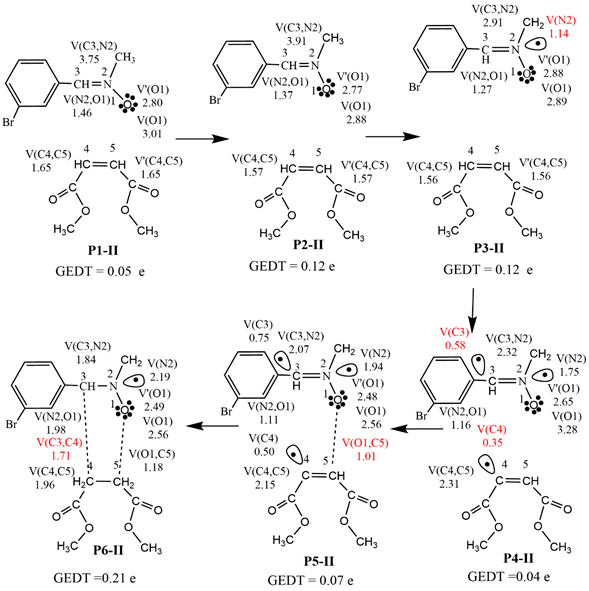
Scheme 3 Sequential bonding changes and most significant valence basin populations in average number of electrons e at the representative IRC points along the endo reaction path of the 32 CA reaction of N-methyl-C-3-bromophenyl-nitrone 1 with dimethyl maleate 2.
In phase V, the formation of the first O1-C5 single bond begins, defined by the Intrinsic Reaction Coordinates (IRC) [57]. point P5-II, at the O1-C5 distance of 1.561 Å, as shown by the formation of disynaptic basin V (O1, C5) integrating 1.01 e. Take note of it at the TS.
Finally, in phase VI, at P6-II, the formation of the second C3-C4 single bond began at a C3-C4 distance of 1.611 Å, as shown by the formation of a disynaptic basin V (C3, C4) integrating 1.71 eV. In cycloadduct 4, the molecular geometry is completely relaxed at O1-C5 and C3-C4 distances of 1.426 and 1.560 Å, respectively. Note that the production of the second O1, C5 starts when the total integrating population of disynaptic basin V (O1, C5) reaches 1.18 e. As a result, the endo reaction route is projected to have a two-stage one-step mechanism [58].
A comparison of the BET values for the endo and exo reaction paths for the 32CA reaction of N-methyl-C-3-bromophenyl-nitrone 1 with dimethyl maleate 2 allows the conclusion of several significant results. The third and fourth phases of the endo reaction path PII result in the formation of the lone pair electron density at N2 nitrogen, as well as the pseudoradical centres at C3 and C4, which are required for the creation of the new C3-C4 and O1-C5 single bonds. At the beginning locations, the total integrating population of the V(C3) and V(C4) basins is 0.58 e, 0.35 e, respectively. While the populations along the exo reaction path PI are V(C3), V(C4) and V(C5) basins are 0.26 e, 0.27 and 0.64 e, respectively. These results suggest that the new C3-C5 and O1-C4 single bonds are being formed at the endo and exo of the same reaction pathway. As a result, GEDT at P2-I (0.13) and P3-I (0.14 e) associated with the exo reaction path increased compared to P2-II (0.12 e) and P3-II (0.12 e) associated with the endo reaction path.
Table 6 ELF valence basin populations of the IRC Structures P1--P6- defining the six phases, characterising the molecular mechanism of the (TS-en) reaction path of 32CA reaction N-methyl-C-3-bromophenyl-nitrone 1 with dimethyl maleate 2. The distances of the forming bond distances are given in angstrom units, Å.
| Phases | I | II | III | IV | V | VI | |
|---|---|---|---|---|---|---|---|
| Structures | P1-II | P2-II | P3-II TS-endo | P4-II | P5-II | P6-II | 4 |
| d(C3-C4) | 2.833 | 2.426 | 2.301 | 2.119 | 1.959 | 1.611 | 1.560 |
| d(O1-C5) | 2.511 | 2.026 | 1.874 | 1.679 | 1.561 | 1.439 | 1.426 |
| GEDT | 0.05 | 0.12 | 0.12 | 0.04 | 0.07 | 0.21 | 0.22 |
| V(O1) | 3.01 | 2.88 | 2.89 | 3.28 | 2.56 | 2.56 | 2.52 |
| V’(O1) | 2.80 | 2.77 | 2.88 | 2.65 | 2.48 | 2.49 | 2.48 |
| V (C3, N2) | 3.75 | 3.91 | 2.91 | 2.32 | 2.07 | 1.84 | 1.82 |
| V (N2, O1) | 1.46 | 1.37 | 1.27 | 1.16 | 1.11 | 0.98 | 0.99 |
| V(N2) | 1.14 | 1.75 | 1.94 | 2.19 | 2.26 | ||
| V (C4, C5) | 1.65 | 1.57 | 1.56 | 2.31 | 2.15 | 1.96 | 1.92 |
| V’ (C4, C5) | 1.65 | 1.57 | 1.56 | ||||
| V(C3) | 0.58 | 0.75 | |||||
| V(C4) | 0.35 | 0.50 | |||||
| V(C5) | |||||||
| V (O1, C5) | 1.01 | 1.18 | 1.23 | ||||
| V (C3, C4) | 1.71 | 1.78 |
Conclusion
The 32CA reactions of N-methyl-C-3-bromophenyl-nitrone 1 with dimethyl maleate 2 were investigated using MEDT at the MPWB95/6-311++G(d,p) level of theory. The ELF topological analysis of N-methyl-C-3-bromophenyl-nitrone 1 shows clearly that nitrone is a zwitterionic TAC participating in zw-type 32CA reactions. N-methyl-C-3-bromophenyl-nitrone 1 and dimethyl maleate 2 undergo a one-step 32CA reaction. The FEDF N-methyl-C-3-bromophenyl-nitrone 1 to dimethyl maleate 2 is predicted, since N-methyl-C-3-bromophenyl-nitrone 1 has a larger electronic chemical potential and more nucleophilicity than dimethyl maleate 2 both the stereochemical pathways of these exergonic 32CA reactions have negative Gibbs free energy. The activation enthalpy for the 32CA reaction leading to the exo cycloadduct 3 is higher than that along the endo reaction path. The geometrical characteristics show that at all TSs, the production of C-O or C-C single bonds has not yet began.
The BET study of the 32CA reaction between N-methyl-C-3-bromophenyl-nitrone 1 and dimethyl maleate 2. The synthesis of 3 demonstrates the formation of non-bonding N2 electron density region at the begin of the reaction path with a GEDT of 0.05 e. As a result, increases in GEDT result in a reduction in the cost of energy along the reaction pathways. The current MEDT analysis enables us to conclude that the increased acceleration for the endo stereoselective zw-type 32CA reaction between N-methyl-C-3-bromophenyl-nitrone 1 and dimethyl maleate 2 passing through TS-en is due to the high polar character of this 32CA reaction of NEDF.

