











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
The discovery of different types of microorganisms has explained the main reasons for various infectious diseases responsible for the most complex health issues of this century. Organisms like bacteria, fungi and viruses are identified to cause serious health hazards globally which may even lead to death [1]. Although a lot of drugs as potent antimicrobial agents have been identified hitherto, the rise of resistant microorganisms or the development of multi drug resistance in pathogens still remain as a major concern worldwide [2]. Hence, the discovery of new drugs with potent anti-microbial activity, particularly against the resistant strains is therefore highly needed to solve this problem [3]. Highly reactive free radicals and oxygen species that are present in the biological systems may abstract hydrogen atom from membrane, lipid, protein, DNA etc. and consequently lead to damages of several biological species and hence can initiate numerous degenerative diseases [4]. The impairments caused by free radicals can lead to aging, cancer, atherosclerosis and some other serious disorders. Therefore, the removal of free radicals from biological system is very important for the sustainability of cellular machinery and for preventing the commencement and propagation of oxidative diseases [5]. The supplementation of antioxidants (Free radical scavengers) is found to be beneficial for avoiding oxidative damages as they have the ability to trap free radical species.
Coumarins are an important class of benzopyrones found in green plants either in free or combined state and display wide spectrum pharmacological activities [6]. Natural coumarins and their derivatives are of great interest due to their widespread biological properties and have attracted many medicinal chemists for further derivatization and screening them as novel therapeutic agents. Coumarins are reported to be active as antibacterial [7], anti-inflammatory [8] and antiviral agents [9] and the various therapeutic applications of coumarin derivatives include photo chemotherapy, anti-tumor therapy and anti-HIV therapy [10]. The coumarin motif is present within the chemical structure of pharmaceutical drugs such as warfarin, acenocoumarol, carbochromen etc. and in antibiotics such as novobiocin, clorobiocin and coumermycin A1 [11,12]. In view of these interesting pharmacological properties, the exploration of natural or synthetic coumarin derivatives has intrigued chemists for decades for their applicability as drugs. On the other hand, 1,2,3-triazoles are found in diverse bioactive compounds and have shown numerous biological potentials like anticancer [13], immunosuppressant [14], antimicrobial [15], antiviral [16], antiallergic [17] and anti-inflammatory activities [18]. The exceptional properties of 1,2,3-triazoles include high dipole character and hydrogen bonding capability and hence can be used as linkers of various molecules. Furthermore, these compounds are highly rigid and stable under acid/base hydrolysis and oxidative/reductive conditions which makes it a metabolically stable heterocyclic ring [19,20]. Several 1,2,3-triazole containing drug molecules are now available in the market (Fig 1) or is in the clinical trials of final stage [21].

Nowadays, the microwave-assisted organic synthesis (MAOS) is rapidly becoming recognized as a valuable tool for facilitating a wide variety of transformations and hence has significantly extended its scope in drug discovery laboratories [22]. It is well documented that the microwave assistance can lead to remarkable rate enhancement with better reproducibility and less side reactions as compared to standard heating methodologies [23]. In the design and development of new drugs, the employment of molecular hybridization strategy which involves the combination of different pharmacophores may lead to compounds with interesting biological profiles. These combined chemical entities recognized and derived from known bioactive molecules possessing different mechanisms of action could be beneficial for various treatments as they possibly offer some advantages in overcoming drug resistance as well as improving their biological potency [24,25].
Since the combination of two pharmacophores on the same scaffold is a well established approach to more potent drugs [24-27], we focused our attention in the synthesis of some pharmacologically relevant coumarin derivatives containing 1,2,3-triazole moiety. Owing to the instability of coumarin nuclei in basic as well as prolonged heating conditions and as a continuation of our ongoing research in the synthesis of some biologically active molecules [28-31], it has been planned to utilize the copper catalyzed Huigsen 1,3 dipolar cycloaddition, commonly known as click chemistry, for the synthesis of various coumarin analogues under microwave irradiation. The synthesis and biological evaluation of 1,2,3-triazole derivatives linked with coumarin moiety by utilizing copper catalyzed azide-alkyne cycloaddition is recently reported in the literature by various research groups [32-35]. However, most of these results were limited to the usage of aromatic azides and a very few benzylic azides, and the reaction times are generally very high (18-24 h) that may lead to various side-products. Moreover, the scope of diverse aliphatic, acyclic and cyclic azides in this area is relatively unexplored and challenging as it is difficult to handle aliphatic azides. Furthermore, aliphatic azides are less reactive and extremely stable in almost all the reaction media and hence it requires prolonged reaction times for complete conversion of starting materials to products. These observations prompted us to optimize the planned synthetic methodology under microwave irradiation as it will be less time consuming and highly efficient with less side products. In this paper, we report a facile, convenient and rapid access for the microwave irradiated synthesis of a series of coumarins linked with 1,2,3-triazoles. The synthesized compounds were screened for their antimicrobial and antioxidant potencies, and the in-silico docking studies of selected compounds against gyrase enzyme has been subsequently investigated.
Experimental
Chemistry
General
All solvents and reagents were obtained from commercial suppliers and used without any further purification unless otherwise noted. Microwave reactions were performed in a single mode Biotage Initiator Microwave Synthesizer and temperature was monitored using infrared. Analytical TLC was performed on pre-coated aluminum sheets of silica (60 F 254 nm) and visualized by short-wave UV light at λ 254. Melting points were determined on an EZ - Melt automated melting point apparatus. 1H NMR (400 or 300 MHz) and 13C NMR (100 or 75 MHz) were recorded on Bruker Avance II spectrometer and chemical shifts were measured in δ (ppm). The following abbreviations are used for the splitting patterns: s for singlet, d for doublet, t for triplet, m for multiplet and br for broad. LC-MS analyses were performed using ESI/APCI, with an ATLANTIS C18 (50 X 4.6 mm-5µm) column and a flow rate of 1.2 mL/min.
Procedure for the synthesis of 4-methyl-7-hydroxy coumarin intermediate (2)
To the weighed quantity of resorcinol (1 equiv.) and ethyl acetoacetate (1.1 equiv.), the ionic liquid [bmim]Cl·2AlCl3 (1.1 equiv.) was added and the reaction mixture was stirred at 30 °C for 20 min. All additions were carried out in an inert atmosphere. The reaction was quenched by adding 6 M HCl in cold conditions. The resultant product was filtered and further purified by column chromatography to obtain the titled compound 2 as off white solid in 88 % yield. mp: 180-182 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.34 (s, 3H, CH3), 6.10 (s, 1H, ArH), 6.69 (d, J=2.3 Hz, 1H, ArH), 6.78 (dd, J=8.7, 2.3 Hz, 1H, ArH), 7.56 (d, J=8.7 Hz, 1H, ArH), 10.48 (bs, 1H, OH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.0, 102.1, 110.2, 112.0, 112.8, 126.5, 153.5, 154.8, 160.2, 161.1. LC-MS: Calculated 176.2, Observed 177.2. Analysis calcd for C10H8O3: C, 68.18, H, 4.58, O, 27.25 %, found: C, 68.21, H, 4.57, O, 27.22 %.
Procedure for the synthesis of 4-methyl-7-(prop-2-ynyloxy)-2H-chromen-2-one intermediate (3)
To the weighed quantity of 4-methyl-7-hydroxy coumarin 2 (1 equiv.) in acetone, were added K2CO3 (3 equiv.) and propargyl bromide (1.2 equiv.) in inert atmosphere and the reaction mixture was stirred at RT for 12 hours. The reaction completion was monitored by TLC and the mixture was poured into ice-cold water with severe stirring. The solution was extracted with ethyl acetate, separated the organic layer, washed with brine, dried with Na2SO4 and distilled under reduced pressure to obtain the titled compound as brown solid in 93 % yield. mp: 139-141 °C; 1H NMR (400 MHz, CDCl3): δ 2.40 (s, 3H, CH3), 2.57 (s, 1H, CH), 4.76 (d, J=2.3 Hz, 2H, OCH2), 6.16 (s, 1H, ArH), 6.92 (d, J = 2.4, 1H, ArH), 6.94 (s, 1H, ArH), 7.52 (d, J = 9.2 Hz, 1H, ArH). 13C NMR (100 MHz, CDCl3): δ 18.7, 56.3, 76.6, 77.6, 102.3, 112.5, 112.8, 114.4, 125.7, 152.5, 155.1, 160.5, 161.2. LC-MS: Calculated 214.2, Observed 215.2. Analysis calcd for C13H10O3: C, 72.89, H, 4.71, O, 22.41 %, found: C, 72.92, H, 4.70, O, 22.38 %.
General procedure for the synthesis of compounds (4a-t)
To the weighed quantity of intermediate 3 (1 equiv.) in t-butanol/water (1:1), were added azide (1.3 equiv.), CuSO4.5H2O (0.1 equiv.) and sodium ascorbate (0.3 equiv.) and the reaction mixture was placed in the microwave and heated for 2-5 min. at 90 °C at 110 W power. The reaction mixture was quenched with water and extracted with DCM, dried in Na2SO4 and distilled in reduced pressure to obtain the crude product. The crude product was further purified by column chromatography and eluted in varying polarities to obtain the titled compounds 4a-t.
7-((1-(2-Methoxycyclopentyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4a)
White solid: mp 168-170 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 1.62-1.67 (m, 1H, CH), 1.76-1.85 (m, 2H, CH2), 1.96-2.09 (m, 2H, CH2), 2.23-2.28 (m, 1H, CH), 2.49 (d, J=1.7 Hz, 3H, CH3), 3.18 (s, 3H, OCH3), 4.00-4.04 (m, 1H, OCH), 4.83-4.88 (m, 1H, NCH), 5.25 (s, 2H, OCH2), 6.22 (d, J=1.0 Hz, 1H, ArH), 7.02-7.05 (dd, J=8.8, 2.5 Hz, 1H, ArH), 7.15 (d, J=2.4 Hz, 1H, ArH), 7.70 (d, J=8.8 Hz, 1H, ArH), 8.39 (s, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.4, 23.5, 26.7, 33.5, 63.5, 65.7, 69.0, 76.1, 111.6, 113.7, 116.1, 124.9, 128.5, 133.9, 146.6, 154.2, 157.0, 160.6, 160.6. LC-MS: Calculated 355.2, Observed 356.2. Analysis calcd for C19H21N3O4: C, 64.21, H, 5.96, N, 11.82 %, found: C, 64.26, H, 5.93, N, 11.81 %.
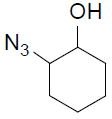
7-((1-(2-Hydroxycyclohexyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4b).
White solid: mp 167-169 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 1.82-1.89 (m, 5H, CH), 1.95-1.97 (m, 3H, CH), 2.49 (d, J=1.7 Hz, 3H, CH3), 3.71-3.73 (m, 1H, OCH), 4.21-4.24 (m, 1H, NCH), 4.95 (d, J=5.9 Hz, 1H, OH), 5.23 (s, 2H, OCH2), 6.22 (d, J=0.9 Hz, 1H, ArH), 7.03-7.06 (dd, J=8.8, 2.4 Hz, 1H, ArH), 7.16 (d, J=2.4 Hz, 1H, ArH), 7.70 (d, J=8.8 Hz, 1H, ArH), 8.25 (s, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.4, 23.4, 24.7, 26.8, 33.5, 61.8, 69.7, 77.1, 112.7, 114.5, 115.8, 125.8, 128.2, 134.0, 147.9, 154.6, 157.6, 160.7, 161.0. LC-MS: Calculated 355.2, Observed 356.2. Analysis calcd for C19H21N3O4: C, 64.21, H, 5.96, N, 11.82 %, found: C, 64.25, H, 5.92, N, 11.81 %.
7-((1-(2-Hydroxycyclopentyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4c).
White solid: mp 155-157 °C; 1H NMR (300 MHz, DMSO-d 6 ): δ 1.96-2.02 (m, 3H, CH), 2.18-2.23 (m, 2H, CH2), 2.25-2.30 (m, 1H, CH2), 2.49 (d, J=1.7 Hz, 3H, CH3), 4.19-4.27 (m, 1H, OCH), 4.58-4.66 (m, 1H, NCH), 5.23 (s, 2H, OCH2), 5.23 (d, J=3.45 Hz, 1H, OH), 6.21 (s, 1H, ArH), 7.01-7.05 (dd, J=8.8, 2.5 Hz, 1H, ArH), 7.15 (d, J=2.5 Hz, 1H, ArH), 7.70 (d, J=8.8 Hz, 1H, ArH), 8.32 (s, 1H, ArH). 13C NMR (75 MHz, DMSO-d 6 ): δ 18.4, 23.4, 26.8, 33.5, 63.8, 69.4, 76.2, 111.9, 113.8, 116.1, 125.8, 128.5, 133.9, 147.7, 154.7, 157.6, 160.8, 160.9. LC-MS: Calculated 341.2, Observed 342.2. Analysis calcd for C18H19N3O4: C, 63.33, H, 5.61, N, 12.31 %, found: C, 63.37, H, 5.59, N, 12.30 %.
7-((1-(2-(2-Phenoxycyclohexyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4d).
White solid: mp 193-195 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 1.72-1.84 (m, 3H, CH), 1.89-1.93 (m, 2H, CH2), 1.99-2.03 (m, 1H, CH), 2.08-2.14 (m, 2H, CH2), 2.46 (d, J=1.2 Hz, 3H, CH3), 3.78-3.84 (m, 1H, OCH), 4.28-4.37 (m, 1H, NCH), 5.28 (s, 2H, OCH2), 6.24 (d, J=1.1 Hz, 1H, ArH), 7.04-7.07 (dd, J=8.2, 2.0 Hz, 1H, ArH), 7.09-7.18 (m, 3H, ArH), 7.17 (d, J=2.5 Hz, 1H, ArH), 7.26-7.41 (m, 2H, ArH), 7.67 (d, J=8.1 Hz, 1H, ArH), 8.39 (s, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.4, 23.4, 24.7, 26.8, 33.5, 64.7, 70.0, 78.1, 112.7, 114.5, 115.8, 118.1, 118.6, 119.2, 125.8, 127.3, 128.2, 129.5, 134.0, 147.9, 154.6, 157.6, 161.8, 160.7, 160.9. LC-MS: Calculated 431.2, Observed 432.2. Analysis calcd for C25H25N3O4: C, 69.59, H, 5.84, N, 9.74 %, found: C, 69.64, H, 5.83, N, 9.72 %.
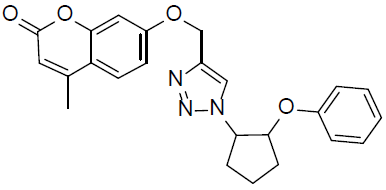
7-((1-(2-(2-Phenoxycyclopentyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4e).
White solid: mp 189-191 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 1.62-1.69 (m, 1H, CH), 1.74-1.86 (m, 2H, CH2), 1.95-2.02 (m, 2H, CH2), 2.17-2.28 (m, 1H, CH), 2.40 (d, J=1.1 Hz, 3H, CH3), 4.27 (q, J=1.6 Hz, 1H, OCH), 4.63 (dt, J=2.3, 1.4 Hz, 1H, NCH), 5.26 (s, 2H, OCH2), 6.28 (d, J=1.2 Hz, 1H, ArH), 7.02-7.04 (dd, J=8.3, 2.1 Hz, 1H, ArH), 7.10-7.19 (m, 3H, ArH), 7.16 (d, J=2.4 Hz, 1H, ArH), 7.27-7.40 (m, 2H, ArH), 7.68 (d, J=8.2 Hz, 1H, ArH), 8.31 (s, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.4, 23.6, 26.8, 33.5, 63.8, 69.7, 75.9, 111.7, 114.9, 116.2, 119.7, 124.8, 125.8, 126.3, 127.7, 128.6, 129.9, 133.8, 147.6, 154.7, 157.4, 159.9, 160.8, 161.2. LC-MS: Calculated 417.2, Observed 418.2. Analysis calcd for C24H23N3O4: C, 69.05, H, 5.55, N, 10.07 %, found: C, 69.08, H, 5.55, N, 10.06 %.
7-((1-(2-(2-Chloropyridin-3-yloxy)cyclohexyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4f).
Light yellow solid: mp 180-182 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 1.70-1.81 (m, 3H, CH), 1.85-1.90 (m, 2H, CH2), 1.95-2.01 (m, 1H, CH), 2.08-2.16 (m, 2H, CH2), 2.45 (d, J=1.1 Hz, 3H, CH3), 3.75-3.82 (m, 1H, OCH), 4.27-4.33 (m, 1H, NCH), 5.27 (s, 2H, OCH2), 6.26 (d, J=1.2 Hz, 1H, ArH), 7.04-7.07 (dd, J=8.2, 1.6 Hz, 1H, ArH), 7.17 (d, J=2.3 Hz, 1H, ArH), 7.41 (d, J=8.0 Hz, 1H, ArH), 7.67 (d, J=8.3 Hz, 1H, ArH), 8.13 (d, J=8.4 Hz, 1H, ArH), 8.39 (s, 1H, ArH), 8.67 (d, J=8.5 Hz, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.4, 23.5, 24.6, 25.9, 32.9, 64.6, 69.0, 77.7, 112.5, 114.4, 115.9, 125.8, 127.4, 128.9, 129.6, 133.8, 145.5, 146.5, 147.9, 154.6, 157.6, 159.8, 160.8, 161.2. LC-MS: Calculated 466.2, Observed 467.2. Analysis calcd for C24H23ClN4O4: C, 61.74, H, 4.97, N, 12.00 %, found: C, 61.78, H, 4.95, N, 11.99 %.
7-((1-(2-(2-Chloropyridin-3-yloxy)cyclopentyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4g).
Light yellow solid: mp 203-205 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 1.90-1.97 (m, 3H, CH), 2.33-2.38 (m, 3H, CH), 2.49 (d, J=1.7 Hz, 3H, CH3), 5.18-5.22 (m, 2H, NCH, OCH), 5.25 (s, 2H, OCH2), 6.21 (d, J=1.0 Hz, 1H, ArH), 7.00-7.03 (dd, J=8.8, 2.4 Hz, 1H, ArH), 7.13 (d, J=2.4 Hz, 1H, ArH), 7.29-7.32 (m, 1H, ArH), 7.47-7.49 (dd, J=8.2, 1.3 Hz, 1H, ArH), 7.68 (d, J=8.8 Hz, 1H, ArH), 7.95-7.96 (dd, J=4.6, 1.4 Hz, 1H, ArH), 8.41 (s, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.4, 23.6, 26.8, 33.5, 63.8, 69.7, 75.9, 111.7, 114.9, 116.2, 126.3, 127.9, 128.6, 129.6, 133.8, 145.7, 147.6, 149.6, 154.7, 157.7, 159.9, 160.9, 161.2. LC-MS: Calculated 452.0, Observed 453.0. Analysis calcd for C23H21ClN4O4: C, 61.00, H, 4.67, N, 12.37 %, found: C, 61.03, H, 4.65, N 12.35 %.
7-((1-(4-Hydroxypyrrolidin-3-yl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4h).
Yellow solid: mp 146-148 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.39 (d, J=1.2 Hz, 3H, CH3), 3.55-3.76 (m, 1H, CH), 3.79-3.89 (m, 1H, CH), 3.92-4.00 (m, 2H, CH2), 5.24 (d, J=1.8 Hz, 1H, OCH), 5.31 (s, 2H, OCH2), 5.39 (d, J=1.0 Hz, 1H, NCH), 5.60 (d, J=1.2 Hz, 1H, OH), 6.23 (d, J=1.0 Hz, 1H, ArH), 7.03-7.06 (dd, J=8.2, 1.5 Hz, 1H, ArH), 7.16 (d, J=2.4 Hz, 1H, ArH), 7.70 (d, J=8.2 Hz, 1H, ArH), 8.55 (s, 1H, ArH), 9.72 (bs, 1H, NH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.8, 44.3, 53.2, 64.8, 69.4, 77.4, 111.9, 113.7, 116.3, 125.7, 129.4, 133.6, 146.9, 154.7, 157.6, 160.6, 160.8. LC-MS: Calculated 342.0, Observed 343.0. Analysis calcd for C17H18N4O4: C, 59.64, H, 5.30, N, 16.37 %, found: C, 59.71, H, 5.26, N, 16.36 %.
7-((1-(4-Phenoxypyrrolidin-3-yl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4i).
Light yellow solid: mp 190-192 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.49 (d, J=1.6 Hz, 3H, CH3), 3.55-3.60 (m, 1H, CH), 3.76-3.79 (m, 1H, CH), 3.89-3.96 (m, 2H, CH), 5.31 (s, 2H, OCH2), 5.40 (d, J=4.0 Hz, 1H, OCH), 5.61 (d, J=4.8 Hz, 1H, NCH), 6.23 (d, J=0.9 Hz, 1H, ArH), 7.00-7.05 (m, 4H, ArH), 7.17 (d, J=2.4 Hz, 1H, ArH), 7.32-7.36 (m, 2H, ArH), 7.71 (d, J=8.8 Hz, 1H, ArH), 8.55 (s, 1H, ArH), 9.72 (bs, 1H, NH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.8, 44.3, 53.2, 64.8, 69.4, 77.4, 111.9, 113.7, 116.3, 119.2, 120.3, 122.5, 125.7, 127.8, 128.6, 129.4, 133.6, 146.9, 154.7, 157.6, 158.8, 159.9, 160.8. LC-MS: Calculated 418.2, Observed 419.2. Analysis calcd for C23H22N4O4: C, 66.02, H, 5.30, N, 13.39 %, found: C, 66.05, H, 5.29, N, 13.39 %.
7-((1-Benzyl-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one [32] (4j).
White solid: mp 118-120 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.49 (d, J=1.2 Hz, 3H, CH3), 4.98 (s, 2H, NCH2), 5.25 (s, 2H, OCH2), 6.24 (d, J=1.2 Hz, 1H, ArH), 7.09-7.11 (dd, J=8.2, 1.6 Hz, 1H, ArH), 7.16-7.19 (dd, J=8.6, 2.1 Hz, 2H, ArH), 7.21 (d, J=2.2 Hz, 1H, ArH), 7.36 (m, 1H, ArH), 7.69-7.71 (dd, J=8.7, 1.92 Hz, 2H, ArH), 7.73 (d, J=7.3 Hz, 1H, ArH), 8.47 (s, 1H, ArH. 13C NMR (100 MHz, DMSO-d 6 ): δ 18.8, 59.5, 76.8, 112.4, 114.5, 116.5, 125.8, 126.7, 128.6, 129.1, 131.6, 134.6, 143.5, 148.9, 154.5, 156.9, 159.3, 160.5. LC-MS: Calculated 347.0, Observed 348.0. Analysis calcd for C20H17N3O3: C, 69.15, H, 4.93, N, 12.10 %, found: C, 69.20, H, 4.91, N, 12.09 %.
7-((1-(4-Fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4k).
Off white solid: mp 175-177 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.47 (d, J=1.1 Hz, 3H, CH3), 4.97 (s, 2H, NCH2), 5.24 (s, 2H, OCH2), 6.22 (d, J=1.0 Hz, 1H, ArH), 7.01-7.04 (dd, J=7.8, 1.8 Hz, 2H, ArH), 7.11-7.14 (dd, J 1=7.9, 1.9 Hz, 1H, ArH), 7.21 (d, J=2.5 Hz, 1H, ArH), 7.55-7.57 (dd, J=8.2, 2.2 Hz, 2H, ArH), 7.70 (d, J=7.4 Hz, 1H, ArH), 8.51 (s, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.8, 59.5, 76.8, 112.4, 114.5, 115.5 & 115.7 (d, 2 J CF =21 Hz), 116.5, 125.8, 128.6, 129.7 & 129.8 (d, 3 J CF =8 Hz), 134.6, 142.8, 148.9, 156.9, 158.3 & 160.7 (d, 1 J CF =241.70 Hz), 161.3, 162.5. LC-MS: Calculated 365.0, Observed 366.0. Analysis calcd for C20H17N3O3: C, 65.75, H, 4.41, N, 11.50 %, found: C, 65.79, H, 4.38, N, 11.51 %.
7-((1-(4-Chlorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one [33] (4l).
White solid: mp 162-163 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.46 (d, J=1.2 Hz, 3H, CH3), 4.95 (s, 2H, NCH2), 5.21 (s, 2H, OCH2), 6.24 (d, J=1.3 Hz, 1H, ArH), 7.03 (dd, J=7.6, 1.4 Hz, 1H, ArH), 7.10-7.13 (dd, J=7.8, 1.8 Hz, 2H, ArH), 7.29 (d, J=2.6 Hz, 1H, ArH), 7.48-7.51 (dd, J=8.1, 2.1 Hz, 2H, ArH), 7.73 (d, J=7.6 Hz, 1H, ArH), 8.43 (s, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.7, 59.7, 76.8, 112.4, 114.5, 116.5, 125.8, 128.6, 129.0, 131.3, 134.6, 138.8, 143.7, 148.9, 154.5, 155.7, 158.2, 160.4. LC-MS: Calculated 381.1, Observed 382.1. Analysis calcd for C20H16ClN3O3: C, 62.91, H, 4.22, N, 11.01 %, found: C, 62.95, H, 4.21, N, 10.99 %.
4-((4-((4-Methyl-2-oxo-2H-chromen-7-yloxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)benzonitrile (4m).
White solid: mp 175-177 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.51 (d, J=1.1 Hz, 3H, CH3), 4.99 (s, 2H, NCH2), 5.29 (s, 2H, OCH2), 6.33 (d, J=1.4 Hz, 1H, ArH), 7.01-7.04 (dd, J=7.6, 1.2 Hz, 1H, ArH), 7.09-7.12 (dd, J=8.0, 2.0 Hz, 2H, ArH), 7.28 (d, J=2.7 Hz, 1H, ArH), 7.41-7.44 (dd, J=8.4, 2.0 Hz, 2H, ArH), 7.77 (d, J=7.6 Hz, 1H, ArH), 8.50 (s, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.7, 58.9, 76.8, 110.2, 112.4, 114.5, 116.5, 118.2, 125.8, 128.6, 128.8, 130.6, 134.6, 142.9, 148.9, 154.5, 156.8, 158.6, 161.1. LC-MS: Calculated 372.1, Observed 373.1. Analysis calcd for C21H16N4O3: C, 67.73, H, 4.33, N, 15.05 %, found: C, 67.78, H, 4.31, N, 15.04 %.
7-((1-(4-Nitrobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one [32] (4n).
Light yellow solid: mp 144-146°C; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.49 (d, J=1.4 Hz, 3H, CH3), 4.97 (s, 2H, NCH2), 5.26 (s, 2H, OCH2), 6.21 (d, J=1.5 Hz, 1H, ArH), 6.96-6.99 (dd, J=7.4, 1.3 Hz, 1H, ArH), 7.07-7.09 (dd, J=8.0, 1.8 Hz, 2H, ArH), 7.26 (d, J=2.8 Hz, 1H, ArH), 7.37-7.40 (dd, J=8.1, 1.9 Hz, 2H, ArH), 7.68 (d, J=7.5 Hz, 1H, ArH), 8.46 (s, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.7, 59.4, 76.8, 112.4, 114.5, 116.5, 124.9 (2 peaks), 125.8, 128.6, 130.8 (2 peaks), 134.6, 142.9, 146.8, 148.9, 154.5, 156.8, 158.5, 160.9. LC-MS: Calculated 392.1, Observed 393.1. Analysis calcd for C20H16N4O5: C, 61.22, H, 4.11, N, 14.28 %, found: C, 61.27, H, 4.08, N, 14.26 %.
7-((1-(4-Hydroxybenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4o).
Light brown solid: mp 144-146 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.46 (d, J=1.1 Hz, 3H, CH3), 4.93 (s, 2H, NCH2), 5.19 (s, 2H, OCH2), 6.24 (d, J=1.3 Hz, 1H, ArH), 6.94-6.97 (dd, J=7.3, 1.4 Hz, 1H, ArH), 7.07-7.10 (dd, J=7.6, 1.5 Hz, 2H, ArH), 7.28 (d, J=2.4 Hz, 1H, ArH), 7.36-7.39 (dd, J=8.0, 1.9 Hz, 2H, ArH), 7.68 (d, J=7.4 Hz, 1H, ArH), 8.57 (s, 1H, ArH), 10.67 (bs, 1H, OH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.8, 59.4, 76.8, 112.4, 114.5, 116.5, 117.5 (2 peaks), 125.8, 128.6, 131.1 (2 peaks), 133.4, 134.6, 148.9, 154.5, 155.8, 157.0, 160.3, 161.0. LC-MS: Calculated 363.1, Observed 364.1. Analysis calcd for C20H17N3O4: C, 66.11, H, 4.72, N, 11.56 %, found: C, 66.17, H, 4.69, N, 11.55 %.
7-((1-(4-Methoxybenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4p).
Off white solid: mp 178-180 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.51 (d, J=1.0 Hz, 3H, CH3), 3.45 (s, 3H, OCH3), 4.99 (s, 2H, NCH2), 5.29 (s, 2H, OCH2), 6.33 (d, J=1.3 Hz, 1H, ArH), 7.01-7.04 (dd, J=7.6, 1.7 Hz, 1H, ArH), 7.09-7.12 (dd, J=7.8, 2.0 Hz, 2H, ArH), 7.28 (d, J=2.2 Hz, 1H, ArH), 7.41-7.43 (dd, J=8.1, 2.0 Hz, 2H, ArH), 7.77 (d, J=7.4 Hz, 1H, ArH), 8.50 (s, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.7, 58.5, 66.6, 76.9, 112.9, 114.7, 116.0, 116.3 (2 peaks), 125.8, 128.6, 130.6 (2 peaks), 133.0, 134.3, 146.4, 148.7, 155.0, 157.0, 160.7, 161.0. LC-MS: Calculated 377.1, Observed 378.1. Analysis calcd for C21H19N3O4: C, 66.83, H, 5.07, N, 11.13 %, found: C, 66.87, H, 5.05, N, 11.11 %.
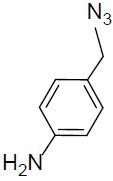
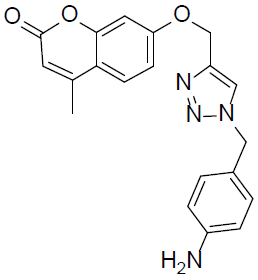
7-((1-(4-Aminobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4q).
Yellow solid: mp 173-175 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.46 (d, J=1.2 Hz, 3H, CH3), 4.95 (s, 2H, NCH2), 5.21 (s, 2H, OCH2), 5.88 (bs, 2H, NH2), 6.24 (d, J=1.4 Hz, 1H, ArH), 7.03-7.05 (dd, J=7.2, 1.5 Hz, 1H, ArH), 7.10-7.13 (dd, J=7.5, 1.8 Hz, 2H, ArH), 7.29 (d, J=2.4 Hz, 1H, ArH), 7.48-7.51 (dd, J=7.8, 2.0 Hz, 2H, ArH), 7.73 (d, J=7.8 Hz, 1H, ArH), 8.43 (s, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.8, 59.4, 76.8, 112.4, 114.5, 116.5, 117.5 (2 peaks), 125.8, 128.6, 129.8, 131.1 (2 peaks), 134.6, 148.9, 154.5, 155.8, 157.0, 160.3, 161.0. LC-MS: Calculated 362.1, Observed 363.1. Analysis calcd for C20H18N4O3: C, 66.29, H, 5.01, N, 15.46 %, found: C, 66.32, H, 5.00, N, 15.44 %.
7-((1-(4-(Methylamino)benzyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4r).
Off white solid: mp 177-179 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.51 (d, J=1.2 Hz, 3H, CH3), 3.02 (s, 3H, NCH3), 4.99 (s, 2H, NCH2), 5.29 (s, 2H, OCH2), 6.33 (d, J=1.2 Hz, 1H, ArH), 7.01-7.04 (dd, J=7.2, 1.3 Hz, 1H, ArH), 7.09-12 (dd, J=7.5, 1.8 Hz, 2H, ArH), 7.28 (d, J=2.7 Hz, 1H, ArH), 7.41-7.43 (dd, J=8.1, 2.2 Hz, 2H, ArH), 7.77 (d, J=7.4 Hz, 1H, ArH), 7.84 (bs, 1H, NH), 8.53 (s, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.6, 38.4, 59.1, 76.2, 111.9, 114.3, 116.1, 117.5 (2 peaks), 126.2, 128.2, 129.7, 130.8 (2 peaks), 134.1, 147.9, 154.1, 155.7, 156.9, 159.1, 160.9. LC-MS: Calculated 376.2, Observed 377.2. Analysis calcd for C21H20N4O3: C, 67.01, H, 5.36, N, 14.88 %, found: C, 67.06, H, 5.34, N, 14.87 %.
4-Methyl-7-((1-(4-methyl-benzyl)-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one (4s).
White solid: mp 171-173 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.33 (s, 3H, CH3), 2.46 (d, J=1.4 Hz, 3H, CH3), 4.93 (s, 2H, NCH2), 5.19 (s, 2H, OCH2), 6.24 (d, J=1.3 Hz, 1H, ArH), 6.94-6.97 (dd, J=7.5, 1.3 Hz, 1H, ArH), 7.07-7.09 (dd, J=7.8, 1.7 Hz, 2H, ArH), 7.28 (d, J=2.5 Hz, 1H, ArH), 7.36-7.39 (dd, J=8.1, 1.88 Hz, 2H, ArH), 7.68 (d, J=7.4 Hz, 1H, ArH), 8.57 (s, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.3, 26.8, 58.7, 76.8, 112.4, 114.5, 116.5, 125.8, 128.1, 128.6 (2 peaks), 130.1 (2 peaks), 134.6, 135.4, 136.3, 148.9, 154.5, 156.9, 159.3, 160.5. LC-MS: Calculated 361.0, Observed 362.0. Analysis calcd for C21H19N3O3: C, 69.79, H, 5.30, N, 11.63 %, found: C, 69.84, H, 5.29, N, 11.61 %.
7-((1-(4-Ethylbenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4t).
White solid: mp 175-177 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.31 (t, J=4.5 Hz, 3H, CH3), 2.44 (d, J=1.2 Hz, 3H, CH3), 4.90 (s, 2H, NCH2), 5.06 (q, J=4.7 Hz, 2H, CH2), 5.19 (s, 2H, OCH2), 6.24 (d, J=1.3 Hz, 1H, ArH), 6.90-6.93 (dd, J=7.3, 1.2 Hz, 1H, ArH), 7.05-7.08 (dd, J=7.7, 1.6 Hz, 2H, ArH), 7.21 (d, J=2.6 Hz, 1H, ArH), 7.37-7.40 (dd, J=8.0, 1.8 Hz, 2H, ArH), 7.66 (d, J=7.5 Hz, 1H, ArH), 8.53 (s, 1H, ArH). 13C NMR (100 MHz, DMSO-d 6 ): δ 18.9, 19.0, 31.1, 58.1, 76.6, 112.3, 115.3, 116.4, 126.1, 128.3, 128.4 (2 peaks), 129.9 (2 peaks), 133.9, 135.0, 135.8, 148.0, 154.3, 155.8, 159.7, 160.9. LC-MS: Calculated 375.2, Observed 376.2. Analysis calcd for C22H21N3O3: C, 70.38, H, 5.64, N, 11.19 %, found: C, 70.43, H, 5.63, N, 11.16 %.
Biology
The experimental procedure for the determination of biological activities is detailed in the Supporting Information.
In silico studies
An entirely in-house developed drug discovery informatics system OSIRIS was used to perform ADMET based calculations. It is a Java based library layer that provides reusable cheminformatics functionality and was used to predict the toxicity risks and overall drug score via in silico [52]. The structure of synthesized molecules and the standards were drawn in ChemBioDraw tool (ChemBioOffice Ultra 14.0 suite) assigned with proper 2D orientation and structure of each one was checked for structural drawing error. Energy of each molecule was minimized using ChemBio3D (ChemBioOffice Ultra 14.0 suite). The energy minimized ligand molecules were then used as input for AutoDockVina, in order to carry out the docking simulation [53]. The protein databank (PDB) coordinate file entitled ‘2XCT.pdb’ was used as receptor (protein) molecule which is a structure of S. aureus gyrase in complex with Ciprofloxacin and DNA [54]. All the water molecules were removed from the receptor and SPDBV DeepView was used to automatically rebuild the missing side chains in the receptor. The Graphical User Interface program ‘MGL Tools’ was used to set the grid box for docking simulations. The grid was set so that it surrounds the region of interest (active site) in the macromolecule.
In the present study, the active site was selected based on the amino acid residues of 2XCT, which are involved in binding with Ciprofloxacin. Therefore, the grid was centered at the region including the 2 amino acid residues (Arg 458 and Gly 459) and 4 nitrogenous bases from DNA that is guanine (G), adenine (A), thymine (T) or cytosine (C) as evidenced by the work of Bax et al., 2010 [55]. This surrounds the active site. The grid box volume was set to 8, 14, and 14 Å for x, y and z dimensions respectively, and the grid center was set to 3.194, 43.143 and 69.977 for x, y and z center respectively, which covered the 2 amino acid residues and 4 nitrogenous bases in the considered active pocket. AutoGrid 4.0 Program supplied with AutoDock 4.0 was used to produce grid maps [55]. The docking algorithm provided with AutoDockVina was used to search for the best docked conformation between ligand and protein. During the docking process, a maximum of 100 conformers was considered for each ligand. All the AutoDock docking runs were performed in Core i7 Intel processor CPU with 8 GB DDR3l RAM. AutoDockVina was compiled and run under Windows 8.0 professional operating system. LigPlot+ [56] and PyMol [51] were used to deduce the pictorial representation of interaction between the ligands and the target protein.
Results and discussion
Chemistry
As depicted in Schemes 1 and 2, we started our synthetic strategy by the synthesis of parent 4-methyl-7-hydroxy coumarin 2 by the modified Pechmann cyclization reaction in which resorcinol 1 was treated with ethyl acetoacetate in 1-butyl-3-methylimidazolium chloro aluminate at 30 °C for 20 minutes [36]. The obtained 4-methyl-7-hydroxy coumarin intermediate 2 was then treated with propargyl bromide in K2CO3 to procure the O-propargylated product 3. The alkyne intermediate 3 thus obtained was further subjected to the copper catalyzed 1,3 dipolar cycloaddition reaction with various azides under microwave irradiation at 90 °C with the intention of synthesizing an array of coumarin derivatives with potent antimicrobial and antioxidant properties.


As a model substrate, we started our initial screening by treating the alkyne intermediate 3 with 2-methoxy cyclopentyl azide in sodium ascorbate and hydrated copper sulfate (CuSO4.5H2O) at 90 °C in microwave. The solvent system used for the reaction optimization was an equimolar mixture of t-butanol and water. To our delight, we identified the reaction completion within 5 min. by TLC and further analysis and purification procured the expected product in 100 % yield (LC-MS) with 97 % isolated yield. In order to validate the predominance of microwave irradiation, we carried out the same reaction at room temperature as well as standard thermal conditions (Table 1, Entry 1). The reaction took 6 h. for completion under conventional heating with 80 % yield while it required 18 h. under ambient temperature and the yield of the product was found to be 86 %.
With the promising results in hand, our next attention was to explore the generality of this synthetic methodology. Keeping this in mind, we treated the intermediate 3 with a series of aliphatic azides under microwave irradiation. Gratifyingly, all the azides reacted efficiently to render the 1,2,3-triazoles linked with coumarins in excellent yields. We also extended our developed methodology for the synthesis of 1,2,3-triazoles with different benzylic substituents linked with coumarins (Table 1, Entries 10-20). To our delight, all the reactions furnished the required products in good to excellent yields. Microwave irradiation proved to be superior in terms of yield as well as reaction time when compared to other standard conditions (Table 1, Entries 1, 2 and 10).
Table 1 Click chemistry reaction of alkyne intermediate 3 with various azides.
| Entry | Azide | Product (4a-t) | Time | Yield b (%) |
|---|---|---|---|---|
| 1 |

|
 4a |
3 min 6 h c 18 h d |
97 80 86 |
| 2 |
|
 4b |
3 min 6 h c 18 h d |
95 76 85 |
| 3 |

|
 4c |
3 min | 94 |
| 4 |

|
 4d |
4 min | 92 |
| 5 |

|
 4e |
4 min | 90 |
| 6 |

|
 4f |
4 min | 93 |
| 7 |

|
 4g |
4 min | 94 |
| 8 |

|
 4h |
3 min | 97 |
| 9 |

|
 4i |
4 min | 90 |
| 10 |

|
 4j |
3 min 6 h c 18 h d |
98 83 87 |
| 11 |

|
 4k |
3 min | 95 |
| 12 |

|
 4l |
3 min | 93 |
| 13 |

|
 4m |
4 min | 92 |
| 14 |

|
 4n |
5 min | 90 |
| 15 |

|
 4o |
3 min | 96 |
| 16 |

|
 4p |
3 min | 95 |
| 17 |
|
 4q |
3 min | 98 |
| 18 |
|
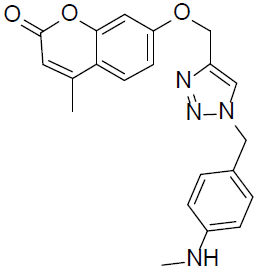 4r |
3 min | 98 |
| 19 |

|
 4s |
3 min | 96 |
| 20 |

|
 4t |
3 min | 95 |
a Reaction conditions: Alkyne 3 (1 equiv.), Azide (1.3 equiv.), CuSO4.5H2O (0.1 equiv.), Sodium ascorbate (0.3 equiv.), t-BuOH/H2O (1:1), microwave irradiated at 90 °C.
b Isolated yield.
c Reaction carried out by conventional heating.
d Reaction carried out at room temperature.
Biology
Antimicrobial activity
The clinical relevance of bacterial and fungal diseases has increased over the past 30 years due to an increasing population of immunocompromised patients who suffer from various illnesses. The development of multi drug resistance among pathogens could be a major reason for this increasing issue [37]. In view of these facts and stimulated by the profound activity profile of coumarins and 1,2,3-triazoles, we carried out the analysis of antibacterial and antifungal activities of the newly synthesized compounds 4a-t against two Gram-positive bacteria (Staphylococcus aureus ATCC 25923 and Bacillus subtilis ATCC 6633), two Gram-negative bacteria (Pseudomonas aeruginosa ATCC 27853 and Escherichia coli ATCC 25922) and three fungi (Aspergillus flavus ATCC 9643, Chrysosporium keratinophilum ATCC90272 and Candida albicans MTCC 227). The results from the evaluation of antimicrobial activities in mg/mL concentration are illustrated in Table 1 and Table 2 of the Supplementary Information. Some of the tested compounds showed promising antibacterial activity when compared to the standard drug, Ciprofloxacin (See Table 1, Supplementary Information). The compounds 4f, 4g, 4k, 4l, 4m and 4s showed promising activity when compared with the standard while the compounds 4b, 4c, 4d, 4j, 4o and 4r failed to show any activity against the tested strains. All the other compounds displayed moderate to poor antibacterial activity.
The minimum inhibitory concentration (MIC) of the more active compounds was determined by broth dilution method (Table 2). From the results, it was acknowledged that S. aureus (5 µg MIC) was the most susceptible, and E. coli (10 µg MIC), P. aeruginosa (10 µg MIC) and B. subtilis (10 µg MIC) were the most insensitive strain among all the bacteria used in this study. The compound 4k was found to be active against all the bacterial strains. The 4a, 4i, 4p (150 µg/mL MIC) and 4t (150 µg/mL MIC) showed weak activity as compared to 4f (10 µg/mL MIC), 4g (10 µg/mL MIC), 4k (10 µg/mL MIC), 4l (100 µg/mL MIC), 4m (100 µg/mL MIC) and 4s (125 µg/mL MIC) against E.coli (Table 2). In the last years, Gram-negative bacteria are frequently being reported to have developed multi drug resistance to many of the antibiotics that are currently available in the market of which E. coli is the most prominent [38,39]. This could be the plausible reason for the high MIC values for E. coli as compared to S. aureus.
Table 2 Minimum inhibitory concentration of synthesized compounds for antibacterial activity.
| Compounds in µg/mL |
Escherichiacoli | Staphylococcusaureus | Pseudomonasaeruginosa | Bacillussubtilis |
|---|---|---|---|---|
| 4a | ––– | 100 | 100 | 50 |
| 4f | 10 | 10 | 10 | 25 |
| 4g | 10 | 10 | 10 | 10 |
| 4i | ––– | 100 | 50 | 100 |
| 4k | 10 | 5 | 10 | 10 |
| 4l | 100 | 100 | 25 | 50 |
| 4m | 100 | 75 | 10 | 25 |
| 4p | 150 | 100 | 75 | 50 |
| 4s | 125 | ––– | 75 | 50 |
| 4t | 150 | 100 | 75 | 100 |
| Ciprofloxacin | 0.6 | 0.2 | 0.5 | 0.4 |
The antifungal activity of the newly synthesized compounds was initially carried out in mill molar concentrations by taking Fluconazole as the standard (See Table 2, Supplementary Information). Among the 20 synthesized organic compounds, only a few compounds inhibited the growth of most of the human pathogenic fungi tested. The fungal sensitivity varied according to the tested species. The compounds showed similar antifungal activities to one another. The minimum inhibitory concentration values for the antifungal activity of more active compounds are summarized in Table 3. Among the tested fungi, C. albicans was less sensitive when compared to the other fungal species. A. flavus and C. keratinophilm showed some differing responses to each organic compound. The compound 4n showed good activity when compared to the remaining compounds. On the other hand, all the newly synthesized organic compounds failed to show good and comparable activity to that of the standard. The compounds 4b, 4c, 4d, 4e, 4h, 4j, 4o and 4r failed to show any activity towards the panel of fungal pathogens.
Table 3 Minimum inhibitory concentration of synthesized compounds for antifungal activity.
| Compounds in µg/mL |
Aspergillus flavus | Chrysosporium keratinophilum | Candida albicans |
|---|---|---|---|
| 4a | 300 | 350 | 400 |
| 4f | 300 | 250 | 350 |
| 4g | >500 | >500 | >500 |
| 4i | >500 | >500 | >500 |
| 4k | 250 | 200 | 200 |
| 4l | >500 | >500 | >500 |
| 4m | >500 | >500 | >500 |
| 4n | 200 | 150 | 150 |
| 4p | >500 | >500 | >500 |
| 4q | >500 | >500 | >500 |
| 4s | 300 | 250 | 350 |
| 4t | 350 | 300 | 400 |
| Fluconazole | 10 | 20 | 30 |
Structure-activity relationships
The presence of electron withdrawing fluoro group in 4k is presumed to be the sole reason for the comparable antibacterial activity of that compound. The presence of heterocyclic ring having a chloro substituent was assumed to be beneficial for the enhanced activity of compounds 4f and 4g. The electron withdrawing groups are expected to increase the lipophilicity and thereby enhance the cell permeability of the molecule and hence improved its potency [40]. In general, it can be summarized that in the present study, the presence of ring substitution with an electron withdrawing group and heterocyclic group at position 1 of 1,2,3-triazoles linked with coumarins is an essential feature for the antimicrobial effect of the synthesized compounds.
Antioxidant activity
The DPPH procedure is one of the most effective methods for evaluating the concentration of radical scavenging materials as it does not have to be generated prior to analysis [41]. DPPH radical scavenging activity evaluation is a rapid and convenient assay for screening the antioxidant activities of products and has been successfully applied for the evaluation of radical scavenging activity of newly synthesized coumarin derivatives [42] as they possess an extended p-conjugated system. Owing to these observations, we directed our work towards the evaluation of antioxidant activity of the synthesized compounds 4a-t by DPPH assay.
The synthesized compounds were subjected to antioxidant screening by taking Butylated hydroxytoluene (BHT) as the standard and our results are summarized in Table 4. In this assay, the standard BHT showed a strong scavenging activity, while the compounds 4b (66.1 %), 4h (74.2 %), 4o (61.8 %), 4q (73.5 %) and 4c (68.3 %) displayed a comparable activity (Fig 2). Unfortunately, the compounds 4g, 4j, 4k, 4p, 4s and 4t didn’t exhibit any activity when compared with the standard and hence are considered as inactive. All the other compounds also showed significant scavenging activity, but demanded higher concentrations of the compounds.
Table 4 Determination of antioxidant activity of the synthesized compounds.
| Entry | Compound | % Inhibition at 100 µg Concentration |
|---|---|---|
| 1 | 4a | 54.2 |
| 2 | 4b | 66.1 |
| 3 | 4c | 68.3 |
| 4 | 4d | 36.4 |
| 5 | 4e | 31.4 |
| 6 | 4f | 26.1 |
| 7 | 4g | –– |
| 8 | 4h | 74.2 |
| 9 | 4i | 41.8 |
| 10 | 4j | –– |
| 11 | 4k | –– |
| 12 | 4l | 28.1 |
| 13 | 4m | 30.7 |
| 14 | 4n | 56.3 |
| 15 | 4o | 61.8 |
| 16 | 4p | –– |
| 17 | 4q | 73.5 |
| 18 | 4r | 29.4 |
| 19 | 4s | –– |
| 20 | 4t | –– |
| 21 | Standard (BHT) | 88.6 |

Structure-activity relationships
The results of antioxidant screening revealed that the presence of electron donating ring systems attached to the position 1 of 1,2,3-triazole ring linked with coumarins is an indispensable characteristic for their radical scavenging activity. The hydrophilic electron-donating groups are expected to facilitate the stabilization of the oxygen-centered radical and reduce the O-H bond dissociation enthalpy (BDE), thereby increasing the radical scavenging activity by hydrogen abstraction [43,44]. This could be the plausible reason for the superior activity of compounds 4b, 4h, 4o, 4q and 4c to that of the other synthesized molecules.
Molecular docking studies
The Gyrase enzyme relieves strain while the double-stranded DNA is being unwound by helicase [45,46]. It is an essential enzyme in all bacteria but absent in higher eukaryotes, hence making it an interesting antibacterial target [47-50]. Furthermore, the mode of antibacterial action of Ciprofloxacin is by significantly inhibiting the gyrase enzyme. Hence, the molecular docking studies of the active compounds with gyrase were carried out and reported. Stimulated by the comparable antibacterial activity of some of the synthesized compounds (4f, 4g, 4k and 4l) with the standard as per the in vivo results, it was thought worthy to substantiate those results by performing the molecular docking studies or in silico studies. The comparative docking of receptor gyrase with 4f, 4g, 4k, 4l and the standard, Ciprofloxacin, exhibited good affinity. They established hydrogen bonding with one or more amino acids in the receptor active pocket as represented in Table 5.
Table 5 Binding affinity (kcal/mol), H-bonds, H-bond length and H-bond formation of the standard and the selected molecules after in silico docking.
| Ligand | Affinity (kcal/mol) |
H-Bonds | H-Bond Length (Å) |
H-Bond Between | Hydrophobic interactions |
|---|---|---|---|---|---|
| 4f | -6.9 | 4 | 2.80 | 4f:N3::Glu435:OE2 | Phe1123, Asp437, Gly436, Gly459, Lys460, His1081 |
| 3.04 | 4f:N :: Gly1082:N | ||||
| 3.09 | 4f:O4 :: Ser1085:OG | ||||
| 3.14 | 4f:N2 :: Glu435:O | ||||
| 4g | -6.3 | 3 | 2.83 | 4g:N4::Glu435:OE2 | Asp512, Ile516, His1081, Gly1082, Arg1122, Phe1123, Gly436 |
| 2.96 | 4g:O3::Asp437:N | ||||
| 3.17 | 4g:O3::Ser438:N | ||||
| 4k | -7.2 | 4 | 2.99 | 4k:O3::Ser1085:OG | Ile516, Lys460, Gly436, Gly459, Phe1123, His1081 |
| 3.12 | 4k:O3::Gly1082:N | ||||
| 3.12 | 4k:N2::Glu435:O | ||||
| 3.15 | 4k:E::Arg1122:NH1 | ||||
| 4l | -6.1 | 1 | 2.90 | 4l:O3::Gly459:N | Arg458, Asp437, Gly436, Arg1122, Phe1123, Glu435, Asp512, Asp510 |
| Ciprofloxacin | -6.2 | 2 | 2.83 | Cipr:O3::Asp510:OD2 | Gly459, Asp437, Gly436, Phe1123, Asp512, |
| 3.02 | Cipr:O2::His1081:ND1 |
The 2D representation of the synthesized ligands 4f, 4g, 4k, 4l and the standard Ciprofloxacin is depicted in Figure 3. The compound 4k was found to be the best of all the molecules taken under investigation as it possessed significant hydrogen bonding as well as hydrophobic interactions. For 4k (Fig 3), hydrophobic contacts were seen with six different residues and four H-bonds were formed with various amino acids (Table 5). The standard Ciprofloxacin (Fig 3) represented hydrophobic contacts with five different residues, later a total of two H-bonds were formed with various amino acids (Table 5). In all the cases of the 2D representation, ligands are highlighted in purple colour. The set of conserved residues that are commonly involved in interaction with the ligands and Ciprofloxacin are encircled with red colour.

Based upon the obtained affinity, the best of the synthesized ligands i.e., 4k along with the standard Ciprofloxacin was subjected to 3D protein-ligand interaction analysis. Figure 4 represents the further extrapolation of binding conformation of 4k and Ciprofloxacin. Figures 4(A) and (B) represent the 3D interaction of 4k and Ciprofloxacin respectively with gyrase by using educational version of PyMol [51]. The ligands are represented in green colour, H-bonds with their respective distances are represented with yellow colour, and the interacting residues are represented in ball and stick model representation.

In the present study, 4k was identified to be the best antibacterial agent among all the synthesized compounds which could be attributed to the electron withdrawing character of fluorine atom as well as the ability of the molecule to form significant hydrogen bonding.
Conclusion
We have achieved a rapid, facile and efficient access for the synthesis of an array of 1,2,3-triazoles linked with coumarins via click chemistry and evaluated their antimicrobial and antioxidant properties. Microwave irradiation proved to be superior to other conventional methods for this synthetic methodology in terms of yield as well as reaction time. The compounds 4k and 4g exhibited promising antibacterial activity when compared with Ciprofloxacin against all the tested bacteria. The in silico docking studies of the more active antibacterials were carried out against the gyrase enzyme and found that 4k possessed significant hydrogen bonding and hydrophobic interactions which could also be the plausible reason for its improved potency along with the presence of electron withdrawing fluoro group. The compound 4n displayed better antifungal activity when compared to other synthesized compounds but were not promising when compared with the standard, flucanazole. The compounds 4h and 4q showed comparable antioxidant activity with the standard, BHT, presumably due to the presence of electron donating substituents. The present study paved the way for the synthesis of various coumarin analogues with significant pharmacological properties and further derivatization and lead optimization are in progress.

