











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
We present evidence of the generation of iodine atoms during the two-electron oxidation of iodide. The detection of intermediates in a two-electron process is of fundamental interest in demonstrating that the multiple electron transfers occur sequentially.[1-3] The electrodeposition of aqueous metal complexes to the crystalline metal phase, M(II)/M(0), is one example that has been addressed in the literature to determine if it occurs sequentially, i.e., through a M(I) intermediate.[1,4] Other studies of two-electron reactions include the reduction of metal complexes[2,5,6] and oxides.[7] In the Sn(IV)/Sn(II) system, a Sn(III) intermediate was detected in Br( media by scanning electrochemical microscopy.[2] The detection of intermediates is of interest because the commonly accepted definition of sequential electron transfer involves the existence of an intermediate.[1,8] Alternatively, Gileadi has proposed that simultaneous two electron transfers can occur with two electrons tunneling simultaneously, i.e., two electron transfers happening within 5 fs.[4] Evans discusses for organic molecules a “bona fide” intermediate that needs to diffuse away from the electrode surface with a minimum distance of escape ≈ 1 nm.[1,9]
In addition to the fundamental importance of the iodide oxidation, this is also a two-electron process of practical importance. The iodide/triiodide couple is of interest because it is an efficient redox mediator in dye-sensitized solar cells, (DSSC).[10] Attempts have been made to find another redox mediator for DSSCs,[11-15] the Iˉ/I3ˉredox couple remains in use. An optimized dye-sensitized solar cell with Iˉ/I3ˉredox couple under one sun and lower irradiance, convert incident photons to electrical current with near unity efficiency at short circuit conditions.[16,17] The electrochemistry of the different iodide species and its implications for DSSCs has been examined by Meyer.[17-21] Including the role of iodine intermediates in the recombination of TiO2 electrons.[20] The metal-to-ligand charge transfer (MLCT) excited state of [Ru(bpz)2(deeb)]2+, where bpz is 2,2-bipyrazine and deeb is 4,4-(CO2CH2CH3)2-2,2-bipyridine, can oxidize iodide (Iˉ) to the iodine atom (I•) in acetonitrile solution.[18] Subsequent reaction of the I• with an iodide anion results in I-I bond formation by generating the iodine radical anion, I2 •ˉ.[18,19] The iodide/triiodide couple has been studied by rotating disk electrode and cyclic voltammetry (CV) in CH3CN with studies performed on Pt [22,23] and Hg.[24] Besides CH3CN, other media have also been studied, e.g., Compton studied the couple in ionic liquids (ref [25] and references therein). While the existence of an intermediate has been postulated from electrochemical kinetic measurements,[22,23,25,26] we are not aware of direct evidence of the existence of a radical intermediate under electrochemical conditions; the evidence described above is for the formation of I2 •ˉ under photoelectrochemical conditions on TiO2. In this paper, we report evidence of the electrochemical generation of I• at F-doped tin oxide (FTO) working electrode. Cyclic voltammetry was used to survey the electrochemical behavior of iodine species, i.e., Iˉ, I3ˉ, and I2 on the FTO working electrode. The radical intermediates of DMPO and TMPO formed after reacting with I• (reaction 1) were characterized using EPR spectroscopy (electron paramagnetic resonance). The products of the radical reaction of I• with tetrabutyl ammonium group and with the solvent were characterized by GC-MS.
Experimental
Reagents
Tetrabutylammonium iodide (TBAI, ≥ 99.0%, Fluka), tetrabutylammonium bromide (TBABr, ≥ 98%, Sigma-Aldrich), tetrabutylammonium perchlorate (TBAP, ≥ 99.0%, Fluka), iodine (≥ 99.99%, Sigma-Aldrich), acetonitrile (≥ 99.8%, Sigma-Aldrich), deuterated chloroform with TMS as received, 5,5-dimethyl-1-pyrroline N-oxide (DMPO , ≥ 97%, Sigma-Aldrich), 3,3,5,5-tetramethyl-1-pyrroline N-oxide (TMPO, 95% Sigma-Aldrich), fluorine doped tin oxide coated glass slides (FTO tec 8, Sigma-Aldrich ), platinum working electrode (dia = 1.6 mm, BASi), carbon rod, silver wire 1.0 mm diameter (99.999%, Alfa Aesar), were purchased. Acetonitrile was purified with activated alumina before using in electrochemical experiments. Alumina powder was used for polishing electrodes (CH Instruments, Austin, TX).
Electrochemical Methods
Tetrabutylammonium iodide (TBAI) was used as the iodide source. A three-compartment electrochemical cell (home made using quartz glass) was used for the experiment. FTO glass slides, a Ag wire, and a carbon rod were used as the working electrode (WE), the quasi-reference electrode (QRE) and the counter electrode (CE) respectively. The reference electrode was calibrated using ferrocene methanol (Eº = 0.15 V vs. SCE) [27] to convert the potentials with respect to the normal hydrogen electrode (NHE). A 0.1 M TBAP in acetonitrile was used as the blank unless otherwise stated. TBAP was purified by recrystallization from ethanol before use. All the electrochemical studies were carried out inside a glove box under Ar atmosphere (O2 = 0.1-0.6 and H2O = 0.6 ppm). A CHI 760D electrochemical station (CH Instruments) was used for all electrochemical measurements. A bulk electrolysis cell consisted of a three-compartment cell with an o-ring to define the surface area of the FTO electrode and two glass frits (medium, Chemglas) to separate the counter electrode and the quasi-reference electrode compartments. The counter electrode was a graphite rod, and a Ag wire was the quasi-reference electrode, calibrated as described above. Fig. 7 below shows the cell used for these experiments and Fig. S9 in the supporting information includes measurements of the cell.
EPR Measurements
Bruker Elexsys E 500 series, X-band spectrometer was used to detect the DMPO and TMPO radical intermediates. The default values for organic radicals were used in the measurements. The microwave power was maintained at 2 mW to avoid saturation, and the scan was traced with a modulation equal to 1 G. The center field [G], sample g-factor and receiver gain [dB] used for the measurement were 3524, 2.000000 and 30 respectively. For ex-situ illumination, a 150 W Xe ozone free arc lamp was used along with an IR filter for the illumination. Shimadzu UV-2550 spectrophotometer was used for spectrophotometric analysis. A Varian 500 MHz NMR was used to characterize the final products. The trapping procedure was: a potential step of ca. 1.9 V vs. NHE was applied to the FTO WE for 30 min. The applied potential was determined by obtaining a CV for the electrolyte before the electrolysis process. The electrolyte contained 1 mM TBAI, 0.1 M TBAP in CH3CN. When TMPO (30 mM) was used as the nitrone spin trap, it was added to the electrolyte before applying the potential step. In the case of DMPO, it was added to the electrolyte after applying the potential for 30 minutes as it was oxidized at the applied potential of 1.9 V vs. NHE. The color of the electrolyte changed from colorless to reddish brown as the electrolyisis progressed, which is an indication of the formation of I3ˉ. The formation of I3ˉ at this potential was confirmed with using UV-Visible spectroscopy that produced the characteristic peaks for the iodide and triiodide species. After applying the potential step, a small aliquot of the electrolyte was then transferred to a quartz capillary tube and subsequently, to a homemade EPR tube with airtight sealing.
Spectroelectrochemical Measurements
Home built polytetrafuran (PTFE) cells with FTO and quartz plates mounted on either side of the beam path were used for these experiments. The solutions were prepared and transferred to the cells in the Ar glove box and closed to perform the experiments without exposing the solutions to air. A CHI 760D electrochemical station was coupled to Shimadzu UV-2550 spectrophotometer to perform in-situ spectroelectrochemical experiments. Two cells were used for the experiments, one as the reference cell and the other as the working sample cell to obtain a background with similar absorbance as the working cell. The potentiostat was connected only to the working cell which contained the reference and the counter electrodes; these were mounted clearing the light beam of the spectrophotometer.
Results and Discussion
Cyclic Voltammograms (CV) of Iodide
The redox behavior of Iˉ was studied at both FTO and Pt. In this case, CVs were obtained for 1 mM iodide in CH3CN with 0.1 M TBAP at FTO and Pt working electrodes. At both electrodes, Iˉ shows two oxidation waves (denoted by W1 and W2 in Fig. 1). W3 and W4 denote the reverse (i.e., reduction waves) of W2 and W1 respectively. The first and second oxidation waves of iodide at Pt are chemically reversible. However, at FTO the waves are less reversible. Fig. 1 a) shows the CVs obtained for (a) 1 mM TBAI/0.1 M TBAP at FTO electrode. For comparison, Fig. 1b) shows the CV for a Pt electrode (note the blank CVs in 0.1 M TBAP in CH3CN).
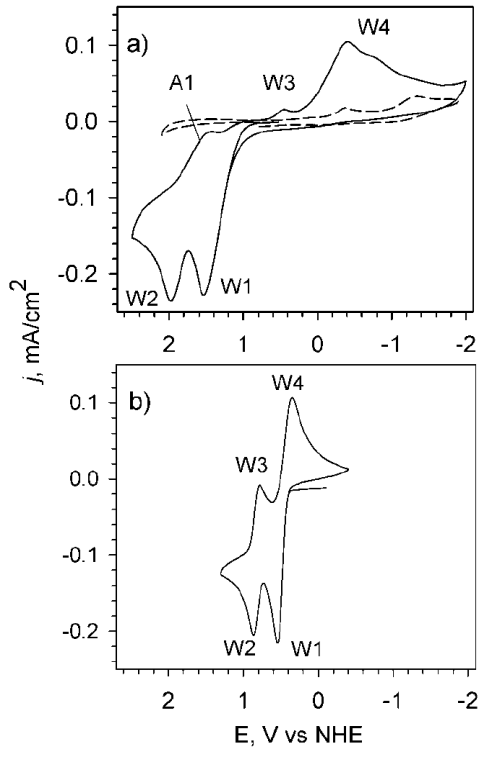
Fig. 1 CVs of 1 mM TBAI + 0.1 M TBAP at a) FTO (diam = 9 mm) and b) Pt (diam = 1.6 mm). (- - - ) Shows the CV of the blank 0.1 m TBAP in CH3CN with small peaks in the cathodic region that are due to FTO background processes. ( = 100 mV/s.
The voltammetry of the oxidation of Iˉ to I2 in acetonitrile at room temperature has been shown to occur in two reversible steps:[22,23]
Fig. 1 a) shows the oxidation of iodide at FTO that also results in two waves similar to the case of Pt (Fig. 1 b)), it is reasonable to assign them to the oxidation reactions given by the equations (2) and (3). To confirm that these oxidation waves are diffusion-limited, we studied the scan rate dependence of these peaks (see supporting information, SI, Fig. S1). The peak anodic currents for both oxidation waves correlate with the square root of scan rate confirming that these are limited by diffusion and not surface confined. Several features are different for the FTO electrode. UV-Visible spectroscopy confirmed the final products of I3 ˉ and I2 (see for example Fig. S8) The oxidations of reactions (2) and (3) occur at significantly larger overpotentials and also the reductions that correspond to the reverse of reactions (2) and (3): the reduction I2/I3 ( (W3) and the reduction of I3 - (W4, note that this reaction requires a catalyst for the fabrication of DSSCs devices). The reduction peak of I2 on FTO (W3 in Fig. 1 a)) is smaller than the other redox waves in part because I2 and Iˉ in CH3CN have a large equilibrium constant [26] to form I3ˉ, K eq = 6 ± 2 × 106 M-1. [21]
During the time it takes to complete the reverse scan of the CV, I2 can react with I- in the bulk of the solution to form additional I3 -. In addition, we point out that the peak current for W3 is proportional to v1/2: at 0.1 V/s is 1.84 x 10-5 while at 0.8 V/s is 9.62 x 10-5 A. Note that this is consistent with our argument because for a completely diffusion-dependent process, without side reactions, the expected current at 0.8 V/s would be proportional to (0.8/0.1)1/2, and therefore, we would expect 5.2 x 10-5 A for 0.8 V/s, much smaller than our observed value. In other words, the experimental data is consistent with W3 being due to byproducts of W2. Additional CVs were performed to validate the peaks on FTO and are shown in the SI (Fig. S2). There is a smaller peak on the reverse of W2 at +1.75 V, (A1) that is assigned to an additional species formed during the oxidation of I- or I3 -. Rowley et al. calculated a Latimer diagram for iodine species in CH3CN;[21] the relevant potentials are given in equations (5)-(8), all written in the form of reductions, all E in V vs. NHE. [21]
Because of the electrochemical potentials listed above, there is no iodide candidate for the byproduct being reduced in A1 at +1.75: there is no iodide species with a potential around 1.7 V that could be generated during the oxidation scan, and later reduced on the reverse peak A1. Therefore, A1 is assigned to a byproduct of the solvent and an intermediate in the oxidation of I- and I3 - that is stable on FTO but not on Pt. Considering the potentials of reactions (5)-(8) and the E 1/2 values for the observed peaks W1 (1.28 V and for W2 ( 1.86 V on FTO (V vs. NHE), it is thermodynamically possible to generate I- and I9 - at the FTO surface when oxidizing of I- and I3 -. However, there is a large overpotential for the oxidation of I2 •- to I2, reaction (5) and therefore, the I2 •- species is less likely to be stable at the FTO surface, and the most likely oxidation reaction is the formation of I•, where the Eº (I•/Iˉ) = 1.23 vs. NHE, reaction (8). In other words, the oxidation of I3 - to I2 on FTO requires large overpotentials that make the formation of I• at the FTO surface a possible parallel path. Therefore, it is possible to generate the radical as a byproduct of the oxidation of I3 - to I2. We note that the diiodide radical anion (I2 •ˉ) formation from I• in excess of Iˉ, has a large rate constant,[19] k = 2.3 × 1010 M-1s-1. Therefore, if iodide is present in solution, the I• radical will quickly produce I2 •ˉ, and this is consistent with our observation that we only see evidence of I• radical under conditions where the local concentration of I- has been depleted. This and other constants previously compiled are shown in Fig. 2.[17]

Fig. 2 Reaction pathways of electrochemically generated I• (adapted from ref 17, Ardo, S; Meyer G., Chem Soc. Rev. 2009, 38, 115)
Trapping of Iodine Atoms
Electrochemically generated I• was trapped using both TMPO and DMPO at FTO working electrode. The large oxidation potential inherent to these nitrone spin traps E0 (DMPO·+/DMPO) ( 1.9 V and E0 (TMPO·+/TMPO) ( 2.0 V vs. NHE)[28] is advantageous in trapping of radicals which forms at higher oxidation potentials. Two different experimental procedures were conducted for the trapping of I• as the oxidation of DMPO competes with the second oxidation of iodide at FTO (see SI, Fig. S3 and compare to Fig. S2). As described in the experimental section, DMPO was added at the later stages of the electrolysis. Note that, ∆E0 for TMPO and DMPO is 100 mV, but at FTO the oxidation peaks are > 400 mV apart due to kinetic differences (Fig. S3). Thus experiments were performed with TMPO present in solution because the oxidation of TMPO on FTO is appreciable at potentials more positive than 2 V vs. NHE. Fig. 3 shows the EPR signal obtained for TMPO and DMPO radical intermediates after reacting with I•. The EPR spectra were recorded at room temperature.

Fig. 3 Spectra in red show the EPR signal for a) DMPO, b) TMPO spin adduct after reacting with I•. Comparison of experimental (red) with simulated spectra (dot-dashed blue line). Blank spectra (black) correspond to the EPR signal obtained without applying a bias to the cell. Solution conditions: 20 mM DMPO (TMPO), 1 mM TBAI, 0.1 M TBAP in CH3CN.
The EPR spectra obtained for the electrolyzed sample of Iˉ with DMPO and TMPO shows a triplet signal with a 1:1:1 intensity ratio. This triplet indicates that the EPR signals originate from an unpaired electron in DMPO or TMPO after reacting with I•, generated by electrolyzing Iˉ at FTO. The unpaired electron should only be affected by the nitrogen atom in those molecules (spin quantum number of the nucleus, I N = 1, so the number of peaks, 2I N +1 = 3).[29]
To further validate the assignment of the EPR spectra, we simulated the
experimental conditions using PEST Winsim Software (National Institute of
Environmental Health Sciences, National Institute of Health, Research Triangle
Park, NC).[30] On the basis of the
simulation, hyperfine coupling constants of 13.65 and 0.68 G for 14N
(1N, I = 1) and 1H (2H, I = 0.5),
respectively, were evaluated for the DMPO radical (Fig. 3 a)). In contrast, a much smaller
hyperfine coupling constant of 6.68 G for 14N (1N, I
= 1) was obtained for the TMPO radical (Fig. 3
b)), which is consistent with the
chemical environment of the unpaired electron. The g-value of
the above two radicals was calculated to be 1.9878 and 1.9913, respectively,
using a value of g = 71.4484
Therefore, by considering the nature of the EPR signal, the following radical intermediates were assigned and shown in Fig. 4 a) and 4b) which describes the observed EPR signal with DMPO and TMPO, consistent with the simulations. The structure in Fig. 4 c) is not detected because hydrogen and iodine with a spin quantum number of the nuclei of 1/2 and 5/2, respectively would introduce more splitting to the EPR signal that what it is observed.
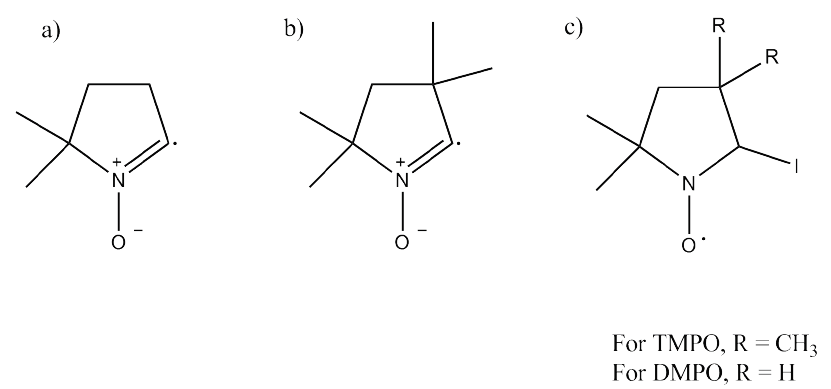
Fig. 4 Structure of the radical intermediate a) DMPO and b) TMPO after reacting with I•. c) Adduct not observed.
Based on the structure of the radical intermediate we propose the mechanism in Fig. 5 for the reaction between electrochemically generated I• with nitrone spin traps. Here, we used TMPO as the model molecule, and the same is true for DMPO.

All the attempts we made to obtain an EPR signal only for I• without adding TMPO or DMPO failed. This is most probably due to the very low concentration of I• formed during the electrochemical process which goes below the detection limit of the EPR instrument and the small lifetime of I• radical in solution. By adding radical traps, we were able to accumulate the organic radical that is significantly more stable as demonstrated by the EPR spectra. The formation of radical intermediate (Fig. 4 a) and b)) instead of nitrone spin adduct was unexpected (Fig. 4 c)). The most probable driving force for the above reaction should be the stable HI acid formation. In all of our experiments with I-, the EPR signal was obtained only after the solution changed from colorless to reddish brown. This can be explained by using the reaction pathways of electrochemically generated I• and their rate constants (Fig. 2).
The electrochemically generated I• can go through two different paths leading ultimately to the formation of I3ˉ as the final product. The fast formation of I2 •ˉ from I• in excess Iˉ, initially competes with the spin trapping reaction (see Fig. 2). Once I3ˉ is accumulated near the surface of the FTO electrode, indicated by the development of reddish brown color of the electrolyte, the concentration of I- ions is depleted due to the formation of I3 -, the rate of formation of I2 •ˉ will be decreased and the spin traps have a higher probability to react with I•.
To further validate the proposed radical intermediate formation mechanism, we conducted the same set of experiments with bromide instead of Iˉ. Since bromine is one row above iodine in the periodic table, we expect it to show similar chemistry to that of iodine. The generation of the radical Br• as an electrochemical intermediate has been demonstrated by SECM in the surface interrogation mode. The detection of the intermediate was further validated by the reaction of Br• with CO: A suspension of Pt black in a Br( solution was bubbled with CO, and the production of CO2 is evidence for the reaction of Br• with CO in solution. As expected, the oxidation of bromide show a similar trend as in Iˉ oxidation except the fact that the peak potentials for each wave appears at higher potentials as shown by Figure S4a. The EPR signal obtained for the Br- experiments (Fig. S4 b)) is consistent with the EPR signal obtained with Iˉ electrolysis. This implies that in both cases (i.e., electrolysis of TBAI and TBABr) they form the same radical intermediate after reacting with TMPO. This is consistent with the formation of either I• or Br• as an intermediate during the electrolysis of Iˉ or Brˉ at FTO working electrode.
Photochemical Generation of Iodine Atoms
We studied the reaction of photogenerated I•with TMPO as a reference system to the method described above. Direct excitation of I2 results in hemolytic dissociation of the I-I bond, yielding two I•, reaction 9, e.g., ref [18]. Therefore, by adding TMPO to a solution containing photogenerated I•, we can study the nature of the reactivity between them. A series of experiments are discussed here, and the data is presented in the SI.
As we expected, an EPR spectrum with the same signature as the spectrum obtained under electrochemical conditions was obtained after the reaction of photogenerated I• with TMPO (Fig. 6). As before, we simulated the EPR spectra, and Fig. 6 shows the comparison with the experimental data. The same hyperfine coupling constant of 6.68 G for α-14N coupling (1N, I = 1) as described in Figure 3b was used, although in this case the g value is slightly shifted from 1.9913 to 1.9888. This simulation results further validate the assignment of the trapped structure, and in turn, our argument on the electrochemical generation of I•. We performed additional studies of the reactivity of the radical in CH3CN without additional electrolyte. The UV Visible spectrum of the solution after 19 h of illumination shows two specific peaks at 291 nm and 361 nm which correspond to I3ˉ in CH3CN,[18] as discussed in the SI (Section V).

Spectroelectrochemical Measurements
A spectroscopic method was implemented to study the formation of iodine species during the electrochemical oxidation of Iˉ at FTO. The details are discussed in the SI, Section VI. The potential step of 1.9 V vs. NHE was applied to the sample cell, and UV visible spectra were recorded simultaneously. The formation of I3ˉ (gives two characteristic peaks at 291 nm and 361 nm), [18] was observed during the electrolysis, and it is shown in Fig. S8. In the relatively short electrolysis time (40 min) the peaks seen are due to I3 - as expected. However, byproducts were formed after longer electrolysis times.
Long-Term Electrolysis
Additional evidence comes from the products of the long-term electrolysis of a TBAI solution. If the intermediate can escape the surface, then we should be able to detect the products of the intermediate with the solvent or electrolyte. Experiments were performed by generating I3 - in an H-type cell with separate compartments for the working, reference and counter electrode. The cell was set up in an Ar glove box to accumulate the products of the reaction. As discussed in the SI (section VII), performing this long-term electrolysis shows an extra peak at around 256 nm that is assigned to a byproduct of the radical reacting with the electrolyte. Therefore, we oxidized of 20 h of a solution and a 0.1 M of TBAI in CH3CN without additional TBAP. The GC-MS analysis is described in detail in the SI (Section VIII) and was performed by comparing several control solutions prepared: 0.1M TBAI in acetonitrile, 0.1M TBAI3, 0.1 M I2 and blank acetonitrile. Some extra peaks were noticed in the GC-MS at retention times; 7.7 min, 8.7 min, 10.2 min, 19.4 min, 21.8 min, 22.1 min, 22.4 min and 28.6 min. We have assigned molecules to the more intense peaks which that appeared in the total ion chromatogram based on their fragmentation patterns and comparing with the NIST database. The data are summarized in Table 1. If the oxidation of Iˉ at FTO is described simply by the two reactions given in equations (2) and (3), then there should not be organic molecules containing iodine such as the ones detected in Table 1. Because these molecules were not detected in any of the controls, a mechanism that involves I- is the most likely explanation for the formation of these organoiodine compounds.

Fig. 7 Picture of the bulk electrolysis cell used for these experiments. The FTO plate was 25 x 25 mm.
Table 1 Summary of compounds assigned based on GC-MS. Data assigned for additional peaks that appear in the total ion chromatogram obtained for the electrolyzed 0.1 M TBAI at FTO.
| Retention Time/min | m/z | Molecular ion/ion | Assigned Molecule |
|---|---|---|---|
| 7.7 | 183.8 | [ICH2CH2CH2CH3]+ | ICH2CH2CH2CH3 Iodobutane |
| 126.7 | I+ | ||
| 71.0 | [CH3CH2CH2CH2CH2]+ | ||
| 57.0 | [CH3CH2CH2CH2]+ | ||
| 8.7 | 129.2 | [NH(CH2CH2CH2CH3)2]+ | NH(CH2CH2CH2CH3)2 N-butylbutan-1-amine |
| 86.1 | [CH3CH2CH2CH2NHCH2]+ | ||
| 57.0 | [CH3CH2CH2CH2]+ | ||
| 44.1 | [CH3CH2NH]+ | ||
| 10.2 | 82.1 | [CH2CH2CH2CH2CN]+ | CH3CH2CH2CH2CN Butyl cyanide |
| 54.0 | [CH2CH2CN]+ | ||
| 40.0 | [CH2CN]+ | ||
| 21.8 | 207.9 | [ICH2CHCHCHCHCH2]+ | ICH2CHCHCHCHCH3 2,4-hexadiene-1-iodo |
| 166.9 | [ICH2CHCH]+ | ||
| 126.9 | I+ | ||
| 81.0 | [CH2CHCHCHCHCH3] | ||
| 40 | [CH2CHCHCHCHCH2]2+ |
We explain the origin of these molecules after the electrolysis of Iˉ at FTO by a radical reaction initiated by the electrochemically generated I•. Again, we point out that these molecules were not detected in the controls, i.e., the same solution conditions but without the electrolysis of I-. The radical I• could react with the tetrabutylammonium ion from the iodide precursor, TBAI. The mechanism in Fig. 8 is proposed as the probable radical initiation reaction, for example, for the formation of iodobutane.

Fig. 8 The proposed mechanism for the reaction between electrochemically generated I• and TBA+ from TBAP leading to the products given in Table 1.
The radical reaction propagates the formed I• and (Bu)3N+• , and this would explain the origin of the products given in Table 1.
Conclusions
Cyclic voltammetry was used to survey the electrochemical behavior of iodine species, i.e., Iˉ, I3ˉ, and I2 on the FTO working electrode. The kinetics of the oxidation of Iˉ, I3ˉ are much slower than on Pt and require substantial overpotentials, and this makes the formation of the I( intermediate thermodynamically stable on the FTO surface. Evidence for a radical comes from radicals of DMPO and TMPO formed after reacting with I• that were characterized using EPR spectroscopy. The radical TMPO• formed in an electrolyzed solution was the same formed during the illumination of I2 to homolytic cleave the I-I bond. The products of the radical reaction of I• with tetrabutyl ammonium group and with the solvent were characterized by GC-MS. Previous reports of the formation of I• and I2 •¯ during the oxidation process of Iˉ by a dye and TiO2 nanoparticles used for DSSCs exist, but to the best of our knowledge this is the first report of evidence of electrochemically generated I•. The oxidation of tetrabutylammonium bromide at FTO also yields similar CVs and generated the same EPR signal corresponding to the trapped radical. Overall, our results indicate that the oxidation of iodide proceeds through the intermediate iodine atoms and thus, it is a sequential two-electron transfer process. Future studies will include attempts to detect the radical near or at the electrode surface or diffusing away from the surface by scanning electrochemically microscopy. Finally, we note the relevance of this report on the possible application of the radical intermediate I( in electrosynthesis, in particular as an electrogenerated reactant or intermediate.[32-34] Given the recent renewed interest in electrosynthesis,[33] the application of this reactant could allow new or optimized transformations. For example, ionic halides are common intermediates in electrochemically driven reactions, and there are reports of Cl(, I(, Br+ and iodium (I+) as electrogenerated reactants, as Frontana-Uribe et al. point out.[32] However, we are not aware of the use of I( radicals, and a setup analogous to the one shown here for bulk electrolysis could allow the application of this radical intermediates. For example, this could be a ‘soft’ oxidizing agent, i.e., a locally generated intermediate that substitutes a strong oxidant[32,33] in potential applications in parallel or convergent electro-synthetic schemes.[34]

