











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
The exponential global economic and population growth has resulted in a rapid depletion of fossil fuel resources and in an increase of environmental concerns that accompany the combustion of these energy supplies.[1] Therefore exploring renewable, sustainable and clean alternative energy sources is fundamental to address these pressing issues.[1] Hydrogen gas produced through hydrogen evolution reactions (HERs) has been considered as a promising alternative fuel due to its high energy density, and its clean and carbon-free nature.[2-4] However, the main challenges of HERs are their high overpotential and sluggish kinetics.[5,6] Thus, to facilitate these reactions, efficient and robust catalysts are required.[7] Although platinum metal has been well demonstrated as the best HER electrocatalyst showing zero overpotential and large current density, its use is highly restricted by its high cost and scarcity.[8] In order to achieve more economically and environmentally viable hydrogen production, significant efforts have been made to explore earth-abundant materials based catalytic systems.[9,10]
Current research on HERs mainly focus on transition metal (Fe, Co, Ni, Mn) and metal-free based electrocatalysts.[9,11-17] Molecular HER electrocatalysts have received substantial interest due to their ease of rational tuning of chemical properties and redox potentials to control their catalytic performance and selectivity.[18-20] In contrast to their bulk heterogeneous counterparts, homogeneous HER molecular catalysts allow for detailed mechanistic study of hydrogen generation by utilizing spectroscopic measurements, among other techniques.[21,22] One of the most frequently studied groups of molecular electrocatalysts are cobalt-nitrogen coordinated complexes such as cobalt diglyoximes, cobalt porphyrins, cobalt salens, cobalt corroles and cobalt chlorins, etc.[23-29] Most of the cobalt complexes studied for hydrogen evolution are supported by electron-withdrawing ligands that tune the redox profile of the metal center by bringing the reduction potential to more anodic ranges, or by redox non-innocent ligands that can participate as electron reservoirs.[30-31] By contrast, cobalt-nitrogen coordinated complexes based HER electrocatalysts with electron-donating ligands are rare. Yet, the use of electron-donating ligands can potentially increase the electron-richness of the cobalt centers and, thus, enhance the reactivity at the catalytic center. Thus, further exploration of cobalt-nitrogen coordinated complexes bearing electron-donating ligands can provide new insight into the development of new ligand architectures that can improve the performance of homogeneous HER electrocatalysts.
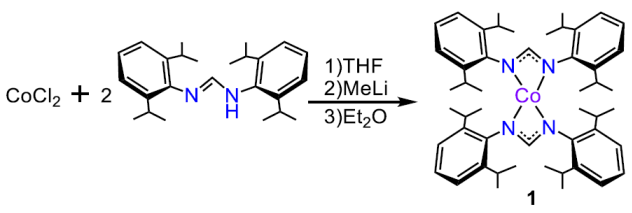
Herein, we report the synthesis of a novel cobalt complex (1) where a single metal center is supported by two electron rich nitrogen-based DippF ligands (DippF = N,N’-bis[2,6-diisopropylphenyl]-formamidinate). We characterized 1 by spectroscopic means including UV-vis, nuclear magnetic resonance (NMR) spectroscopies and X-ray crystallography. Complex 1 was assessed as an electrocatalyst for hydrogen gas evolution by electrochemical measurements in MeCN with three different organic acids, namely p-toluenesulfonic (tosic) acid, benzoic acid and phenol. HER kinetic studies were also performed by foot-of-the-wave analysis (FOWA) in order to provide insights of the relationship between rate constant and acid concentration.
Experimental
General considerations
Synthesis of 1 was accomplished using standard Schlenk techniques under N2 atmosphere and a dry N2-filled glovebox. All solvents used were dried using a Pure Process Technology solvent purification system.
Materials
All materials used were purchased from commercially available sources. Tetrabutylammonium hexafluorophosphate (TBAPF6), p-toluenesulfonic (tosic) acid, benzoic acid, phenol and CoCl2·6H2O were dried under vacuum at 60 °C prior to use. The remaining chemicals were used as received. HDippF was synthesized according to a previously reported method.[32]
Physical measurements
1H NMR spectroscopy was recorded on a Bruker 400 MHz NMR spectrometer, using deuterated benzene (C6D6) as the solvent. The residual protic signal of C6D6 was used as the internal standard (δ=7.16 ppm). UV-vis spectra were obtained using a SEC2000 instrument with a VISUAL SPECTRA 2.1 software.
Information of X-ray crystal structure of 1
A specimen of 1 was used for the X-ray crystallographic analysis. The X-ray intensity data were measured on a Bruker SMART APEX CCD system equipped with a graphite monochromator and a MoKα fine-focus tube (λ = 0.71073 Å).The total exposure time was 4.00 hours. The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. The integration of the data using an orthorhombic unit cell yielded a total of 22716 reflections to a maximum θ angle of 26.00° (0.81 Å resolution), of which 4567 were independent (average redundancy 4.974, completeness = 100.0%, Rint = 3.81%, Rsig = 3.65%) and 4362 (95.51%) were greater than 2σ(F2). The final cell constants of a = 14.4164(7) Å, b = 20.1178(10) Å, c = 15.9876(8) Å, volume = 4636.8(4) Å3, are based upon the refinement of the XYZ-centroids of 9910 reflections above 20 σ(I) with 4.784° < 2θ < 57.66°. Data were corrected for absorption effects using the multi-scan method (SADABS). The ratio of minimum to maximum apparent transmission was 0.889.
The structure was solved and refined using the Bruker SHELXTL Software Package, using the space group C 2 2 21, with Z = 4 for the formula unit, C50H68CoN4. The final anisotropic full-matrix least-squares refinement on F2 with 278 variables converged at R1 = 5.46%, for the observed data and wR2 = 15.32% for all data. The goodness-of-fit was 1.088. The largest peak in the final difference electron density synthesis was 0.486 e-/Å3 and the largest hole was -0.612 e-/Å3 with an RMS deviation of 0.079 e-/Å3. On the basis of the final model, the calculated density was 1.123 g/cm3 and F(000), 1692 e-.
Table 1 Data collection and structure refinement for 1.
| Diffractometer | Bruker SMART APEX CCD | |
| Radiation source | fine-focus tube, MoKα | |
| Theta range for data collection | 1.74 to 26.00° | |
| Index ranges | -17<=h<=11, -24<=k<=24, -19<=l<=19 | |
| Reflections collected | 22716 | |
| Independent reflections | 4567 [R(int) = 0.0381] | |
| Coverage of independent reflections | 100.0% | |
| Absorption correction | multi-scan | |
| Structure solution technique | direct methods | |
| Structure solution program | SHELXS-97(Sheldrick 2008) | |
| Refinement method | Full-matrix least-squares on F2 | |
| Refinement program | SHELXL-2014/6 (Sheldrick, 2014) | |
| Function minimized | Σ w(Fo 2 - Fc 2)2 | |
| Data / restraints / parameters | 4567 / 43 / 278 | |
| Goodness-of-fit on F2 | 1.088 | |
| Final R indices | 4362 data; I>2σ(I) | R1 = 0.0546, wR2 = 0.1517 |
| all data | R1 = 0.0566, wR2 = 0.1532 | |
| Weighting scheme | w=1/[σ2(Fo 2)+(0.0881P)2+7.7217P] where P=(Fo 2+2Fc 2)/3 | |
| Absolute structure parameter | 0.0(0) | |
| Largest diff. peak and hole | 0.486 and -0.612 eÅ-3 | |
| R.M.S. deviation from mean | 0.079 eÅ-3 | |
Evans method
The magnetic susceptibility was measured through 1H NMR spectroscopy, which was carried out with a sealed capillary tube containing pure C6D6 placed inside an NMR tube already loaded with a solution of 1 (0.0160 g) in C6D6 (1.5 mL) in order to measure the chemical shift difference of the solvent caused by the different electronic environments. The number of unpaired electrons (n) was calculated as shown below:[33,34]
where,
The molar susceptibility (
where,
The number of unpaired electrons (n) can be finally calculated through:
Synthesis of 1
Synthesis of 1 was performed in a dried N2-filled glovebox. Anhydrous CoCl2 (0.500 g, 3.85 mmol) was dissolved in 20 mL THF using a 50 mL round-bottom flask. Then, HDippF (2.80 g, 7.70 mmol) was added to the solution. Subsequently, methyllithium (4.81 mL, 7.70 mmol) was added using a glass syringe. The solution changed from a sky blue to a dark aqua-green color. The resulting solution was allowed to vigorously stir under room temperature for three hours. Afterwards, diethyl ether (Et2O) was added to precipitate the LiCl byproduct. The resulting suspension was filtered using a medium coarse glass frit packed with Celite. The filtrate was dried under reduced pressure to obtain 1.058 g of the final green product. The product was crystallized using of a concentrated solution of Et2O. Yield 35.05%. UV-vis: λ max: 564, 628, and 674 nm.
Electrochemical measurements
Electrochemical measurements were recorded on a CHI760D potentiostat using a three-electrode electrochemical cell in a dry N2-filled glovebox, with glassy carbon (4 mm diameter) as the working electrode, platinum wire and Ag/Ag+ as the counter and reference electrode, respectively. Ferrocene (Fc) was added subsequently after each measurement as an internal reference and all potentials displayed are referred to ferrocene/ferrocenium (Fc/Fc+) redox couple.
Hydrogen gas detection
Hydrogen gas was confirmed and quantified using a Gas Chromatograph (GC) after performing bulk electrolysis. Bulk electrolysis was operated using a sealed custom-built three-electrode electrochemical cell with an H shape. The working electrode (1 cm2 carbon rod), reference electrode (Ag/Ag+) were separated with the counter electrode (Pt mesh) through a glass frit. Solutions containing 50 mL of 1 mM of 1, 0.1 M of TBAPF6 and 10 mM of the respective acid studied (tosic acid, benzoic acid, phenol) in MeCN were used for bulk electrolysis at a constant potential of -2.0 V vs. Fc/Fc+ for an hour. An aliquot of 300 µL of the gas in the headspace in the side of the cell containing the working electrode was injected to the GC for analysis.
Calculation of 𝑘𝑜𝑏s and 𝑘 cat from foot-of-the-wave analysis (FOWA)
The HER kinetics can be measured by the foot-of-the-wave analysis (FOWA), which has been developed by Savéant and coworkers.[35] For an ECEC or ECCE mechanistic process,[35-36] the current is described by:
where, F is the Faraday constant (96485 C/mol), A is the surface area of the working electrode (cm2),
According to the Randles-Sevcik equation, the peak current is:[37]
where,
By diving Eq.7 with Eq.8, we can get:
After plotting
Thus
Then the catalytic rate constant (𝑘 cat ) is calculated from:
Calculation of structural geometry index
To disambiguate whether 1 bears closer structural resemblance to a square planar or tetrahedral complex, the model proposed by Yang et al. (Eq. 13) was used.38 In this equation, where
Results and discussion
The solid-state structure of 1 was confirmed by X-ray crystallography (Fig. 1) and the detailed crystallographic data are summarized in Table 2. The bond distances of Co(1)-N(1) and Co(1)-N(2) are 1.998(3) Å and 1.999(3) Å, respectively, which are longer than those of the reported Co diglyoxime complex (1.886(5) Å).[23] The N2-Co1-N2, N1-Co1-N1, N2-Co1-N1 bond angles are found to be 155.0º (2), 151.0º (2), and 119.66º (14) respectively. Based on the bond angles, the calculated geometry index
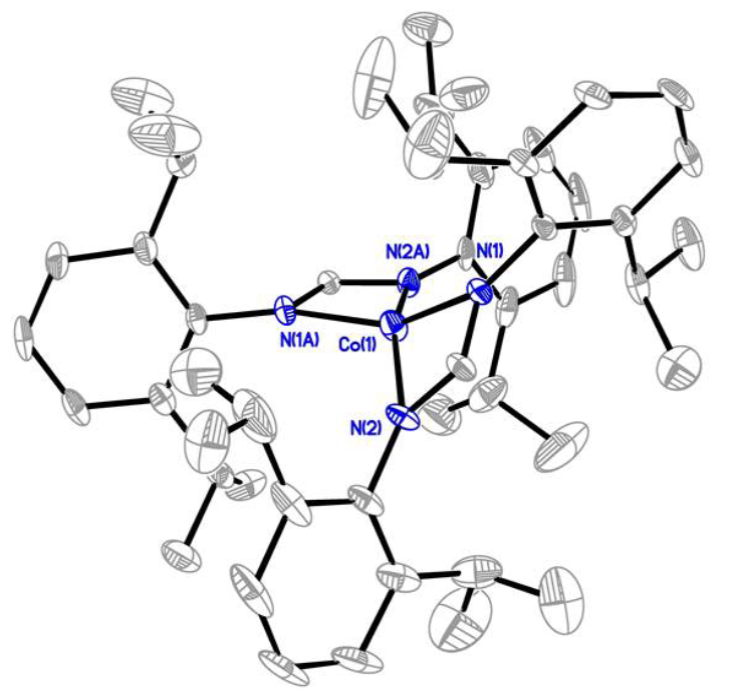
Fig. 1 Solid-state crystal structure of 1 with thermal ellipsoid shown to be 50%. Hydrogen atoms were omitted for clarity. R(int)= 8.58%, R(sigma)= 2.95%.
Table 2 Sample and crystal data for 1.
| Identification code | Co(DippF)2 | ||
| Chemical formula | C50H68CoN4 | ||
| Formula weight | 784.01 g/mol | ||
| Temperature | 100(2) K | ||
| Wavelength | 0.71073 Å | ||
| Crystal system | orthorhombic | ||
| Space group | C 2 2 21 | ||
| Unit cell dimensions | a = 14.4164(7) Å | α = 90° | |
| b = 20.1178(10) Å | β = 90° | ||
| c = 15.9876(8) Å | γ = 90° | ||
| Volume | 4636.8(4) Å3 | ||
| Z | 4 | ||
| Density (calculated) | 1.123 g/cm3 | ||
| Absorption coefficient | 0.406 mm-1 | ||
| F(000) | 1692 | ||
Characterization of 1 by 1H NMR proved challenging due to the intense paramagnetic broadening of the observed signals, reflecting the paramagnetic nature of the complex, and consistent with previous reports of 4-coordinate d7 Co(II) complexes.[39,40] The magnetic susceptibility of 1 was probed through the Evans method. The 1H NMR signal for benzene is shifted by +0.51 ppm in the presence of 1, resulting in a μ eff of 4.13 corresponding to three unpaired electrons (vide supra). This is consistent with other 4-coordinate cobalt complexes bearing nitrogen-donor ligands.[41] The higher μ eff value compared to the predicted spin-only μ eff of 3.87 is attributed to the typical spin-orbit coupling experienced by cobalt.[42,43]
Cyclic voltammograms (CVs) of 1 are presented in Figures 2-4. Compound 1 shows one nonreversible one-electron reduction wave at Ep = -1.59 V vs. Fc/Fc+, resulting from the CoII/CoI reduction couple. The nonreversibility of this event can be attributed to the electron-rich nature of 1. Upon addition of tosic acid (pKa= 8.5 in MeCN), a catalytic wave is observed with an onset potential of -1.2 V vs. Fc/Fc+, accompanying with another reduction wave at Ep = -1.73 V vs. Fc/Fc+. Another two significantly less acidic proton sources, namely benzoic acid (pKa= 21.51 in MeCN) and phenol (pKa= 29.14 in MeCN), were also introduced to evaluate the electrocatalytic activity of 1. The CVs of 1 upon addition of both benzoic acid and phenol show considerable increase of cathodic catalytic current for proton reduction, with an onset potential of -1.45 V and -1.89 V vs. Fc/Fc+ for benzoic acid and phenol, respectively Figures (3-4). The ability of 1 to catalyze proton reduction in weak acidic media, which is noteworthy for this type of study, can be attributed to the enhanced electro-activity of the cobalt center due to the electron-donating formamidinate ligand. Hydrogen gas detection and quantification were carried out after performing bulk electrolysis for an hour at a constant potential of -2.0 V vs. Fc/Fc+ using 1 mM of 1 and 10 mM of the chosen acids. The accumulated charge over time (Fig. 5) shows 36.16, 14.26, 8.08 C of the passed charge for an hour of electrolysis when using tosic acid, benzoic acid and phenol, respectively, corresponding to Faradaic efficiencies of 91.4%, 85.0% and 100% for hydrogen production.
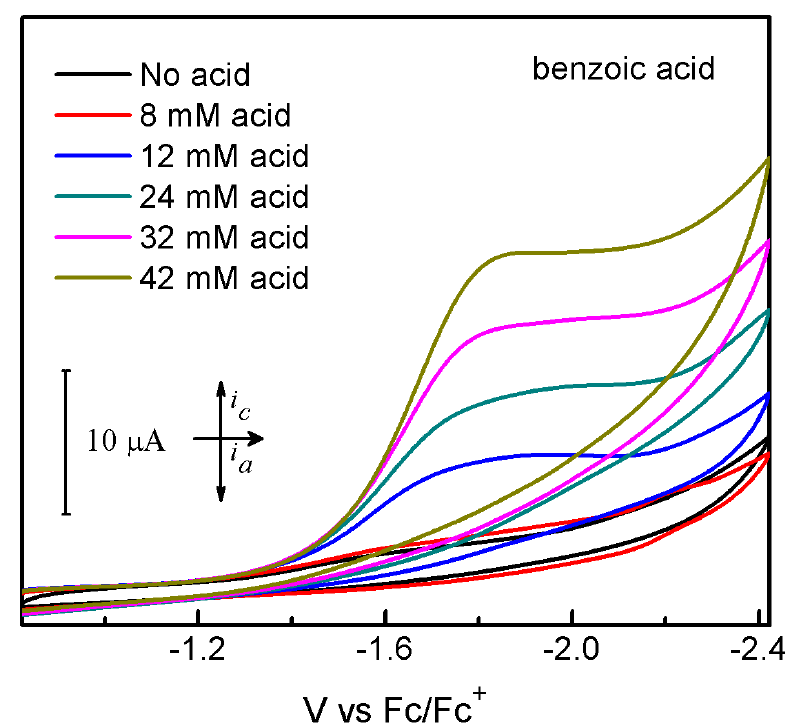
Fig. 2 Cyclic voltammograms of 1 mM 1 without acid (black line) and with tosic acid (colored lines) in MeCN solution containing 0.1 M TBAPF6. Scan rate: 100 mV/s; glassy carbon electrode (4 mm diameter).
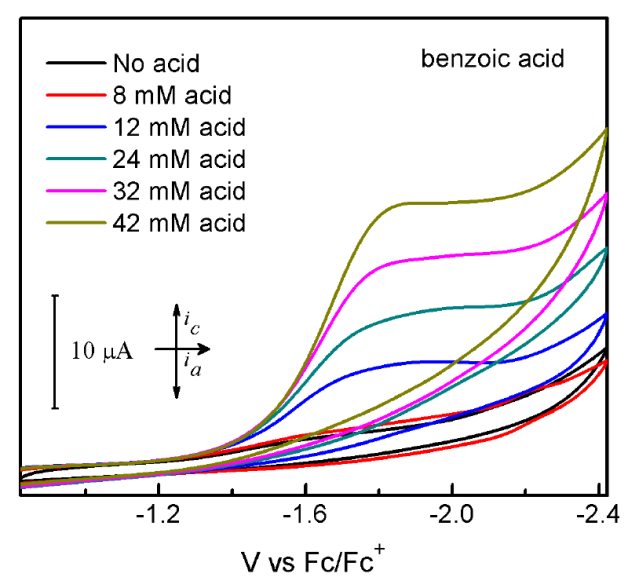
Fig. 3 Cyclic voltammograms of 1 mM 1 without acid (black line) and with benzoic acid (colored lines) in MeCN solution containing 0.1 M TBAPF6. Scan rate: 100 mV/s; glassy carbon electrode (4 mm diameter).
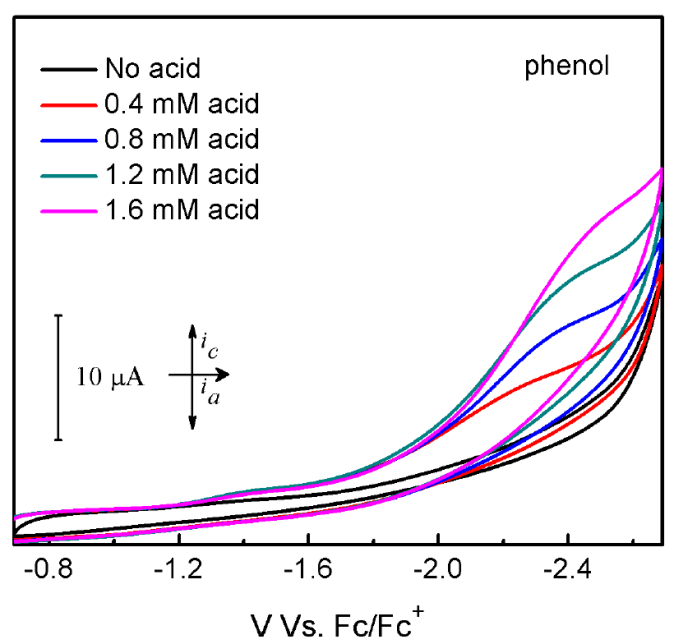
Fig. 4 Cyclic voltammograms of 1 mM 1 without acid (black line) and with phenol (colored lines) in MeCN solution containing 0.1 M TBAPF6. Scan rate: 100 mV/s; glassy carbon electrode (4 mm diameter).
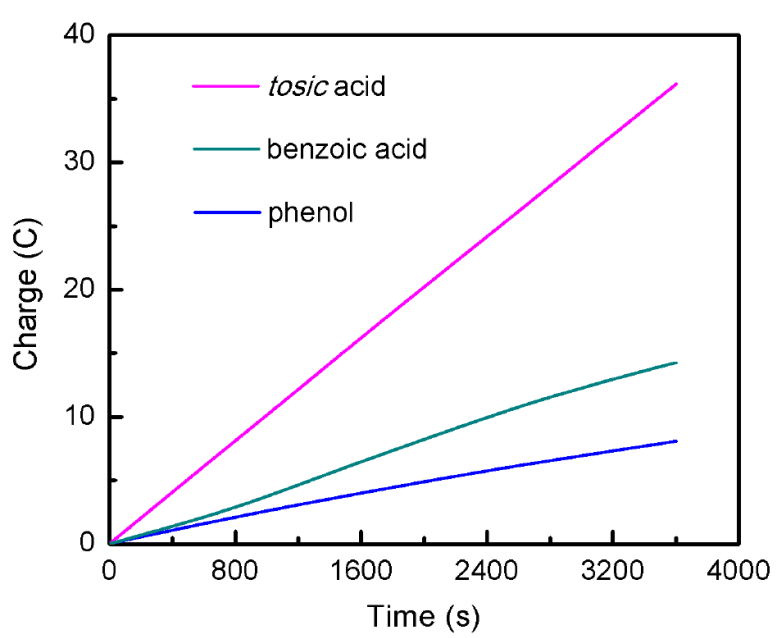
Fig. 5 Bulk electrolysis experiments of 1 mM 1 containing 10 mM of tosic acid, benzoic acid and phenol, respectively. Applied potential: - 2.0 V vs. Fc/Fc+.
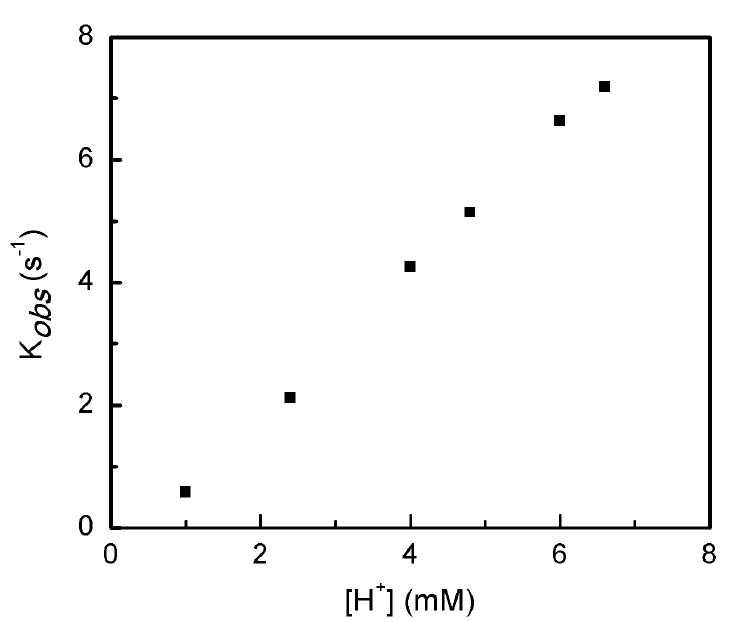
The reaction kinetics of HER using 1 with tosic acid was studied through the use of foot-of-the-wave analysis (FOWA). The FOWA plot is presented in Fig. 6 (a), which is derived from the linear sweep voltammograms shown in Fig. 6 (b). The observed rate constants (𝑘𝑜𝑏s) and catalytic rate constants (𝑘cat) at different concentrations of acid addition are summarized in Table 3 (see experimental section for detailed calculation). The relationship of 𝑘𝑜𝑏s with acid concentration is linear (Fig. 7), suggesting there is a first order dependence on acid concentration.
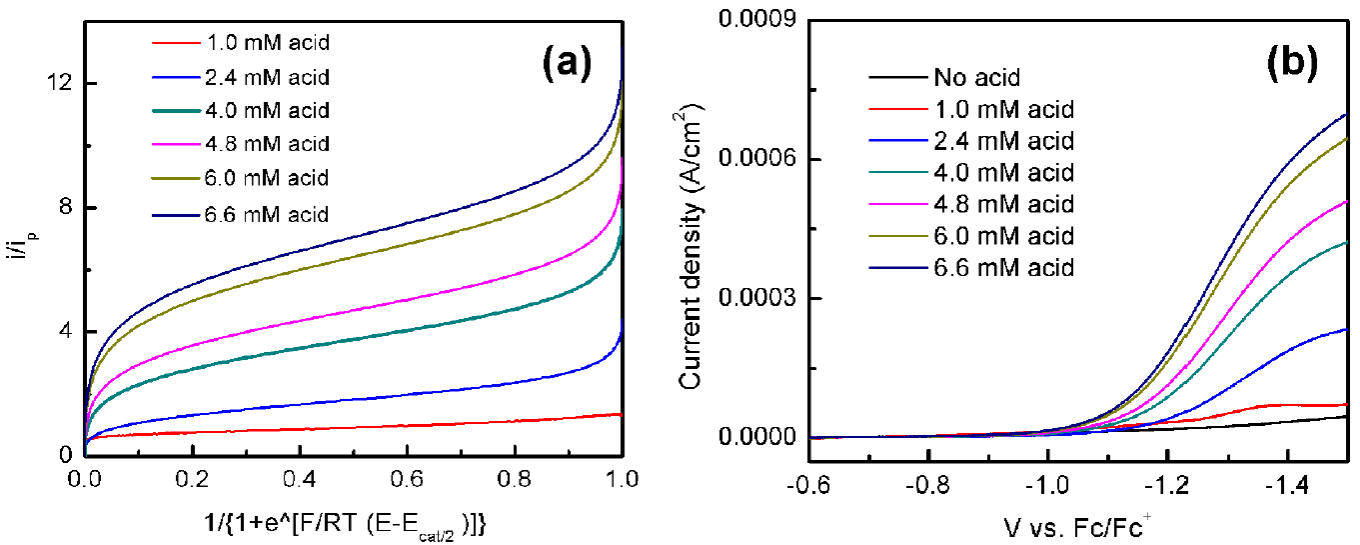
Fig. 6 (a) FOWA plots of 1 mM of 1 at different concentration of tosic acid. Scan rate: 100 mV/s; (b) Linear sweep voltammograms of 1 in MeCN with 0.1 M TBAPF6 titration with tosic acid.
Table 3 Summary of the results of FOWA slope (m), 𝑘𝑜𝑏s and 𝑘 cat at different acid concentration.
| [H+] (mM) | M | 𝑘𝑜𝑏s (s-1) | 𝑘cat (M-1s-1) |
|---|---|---|---|
| 1.0 | 0.58 | 0.0652 | 65.2 |
| 2.4 | 2.12 | 0.872 | 363 |
| 4.0 | 4.25 | 3.50 | 875 |
| 4.8 | 5.14 | 5.12 | 1066 |
| 6.0 | 6.63 | 8.52 | 1420 |
| 6.6 | 7.19 | 10.02 | 1518 |
Conclusion
In conclusion, we have successfully synthesized a cobalt complex anchored by formamidinate ligand. Structural analysis by X-ray crystallography shows that 1 has a distorted square planar chemical structure and magnetic susceptibility measurements show a μ eff of 4.13 B.M. at room temperature, indicative of an electronic structure bearing three unpaired electrons. This molecule is found to be active for electrochemical hydrogen generation with both strong acid and weak acid substrates in MeCN. The electrochemical onset potentials for hydrogen generation range from -1.2 V to -1.89 V vs. Fc/Fc+. This work shows the potential of using electron rich ligand architectures for tuning the redox activity of cobalt HER electrocatalysts.

