











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
The Polyphagous Shot Hole Borer (Coleptera: Curculeonidae: Scolytinae), Euwallacea sp., is an ambrosia beetle native from Asia that has been introduced into Israel, California, South Africa and most recently in México. [1-3] The beetle maintains a symbiotic relationship with Fusarium euwallaceae that it vectors between host trees by carrying spores within a mandibular mycangium.[3,4] This ambrosial fungi is inoculated into host trees and unfortunately it is a virulent pathogen that is responsible of the dieback disease in more than 337 tree species, including agricultural crops and urban forest.[3] To the best of our knowledge, management is currently focused on monitoring, sanitation and direct control using contact or systemic insecticides[5] and chemical control using fungicides are still subject of ongoing investigations.[3] In this context, 1,2,4-triazole derivatives are an important class of heterocyclic compounds with potent pesticidal,[6] herbicidal,[7] and antifungal activities, such as propiconazole, tebuconazol, cyproconazole, and metconazole and the structure unit “(1H-1,2,4-triazol-1-yl)ethanol” is key to their bioactivities (Fig. 1).[8]

All these compounds represent the most important category of fungicides to date, nonetheless, broad use of them has caused severe drug resistance.[9] On the other hand, the modification of the structures of biologically active compounds by means of hydrogen atom substitution by fluorine atom(s) or fluorinated group(s) often leads to an increase in their biological activity and selectivity.[10] These huge successes of fluorinated-containing bioactive compounds continue to stimulate research on fluorine in several areas for drug discovery. For instance, the fungicides developed by Minoru et al., which contain the p-trifluoromethylphenyl moiety, were studied as antifungal agents on mamals [10].
Bioisosterism[11] is an effective strategy for molecular modification and the rational design of bioactive compounds. In this context, we envisioned that 1,2,4-triazoles containing para-trifluormethylphenyl derivatives could be valuable bioactive compounds (Fig. 2).

All the compounds were synthesized and unequivocally characterized by Nuclear Magnetic Resonance 1H, 13C, and DEPTQ 135 spectroscopy and HRMS-QTOF. The biological activity against Fusarium solani and Fusarium euwallaceae were tested and the results showed that most of the synthesized compounds exhibited from modest to good antifungal activities at 1mM concentration when compared to commercially available propiconazol. To further amplify the structure-activity relationship (SAR) of 18 and the resulting activity, the study was focused on varying the substituents R1, R2 and R3 while retaining the para-trifluoromethylphenyl moiety. In this article, we described the synthesis and antifungal activities of some novel triazol derivatives containing a fluorinated group.
Materials and methods
Chemicals. All the chemicals were purchased from Sigma-Aldrich Trading Co. Ltd. Other reagents and solvents were obtained locally and purified according to standard methods before use. All the mixture sensitive reactions were performed under nitrogen atmosphere in glove box. Thin layer chromatography (TLC) analyses were performed on silica gel 60 F254 plates (Merck) and visualization was carried out with p-anisaldehyde, ammonium molybdate, iodine vapors and UV light.
Fungi. The plant pathogenic fungi Fusarium solani were provided by the Phytopathology and Molecular Biology lab at the Clúster Científico y Tecnológico BioMimic(, México. The fungi were grown on potato dextrose agar (PDA) plates at 25 ˚C and maintained at 29 ˚C with periodic subculturing.
Instruments. 1H, 13C, and DEPTQ 135, Nuclear Magnetic Resonance spectra (NMR) were performed on a Bruker Avance III HD 500 spectrometer using a CDCl3 solution with TMS as internal standard. Chemical shifts values (δ) and coupling constants (J) are given in parts per million and Hz, respectively. High Resolution Mass Spectra (HRMS) were obtained in a Q-TOF mass spectrometer equipped with an electrospray ionization (ESI) interface Synapt G2-Si, Waters Inc. Melting points were determined on a Stuart SMP10 apparatus using open glass capillaries and the values are uncorrected.
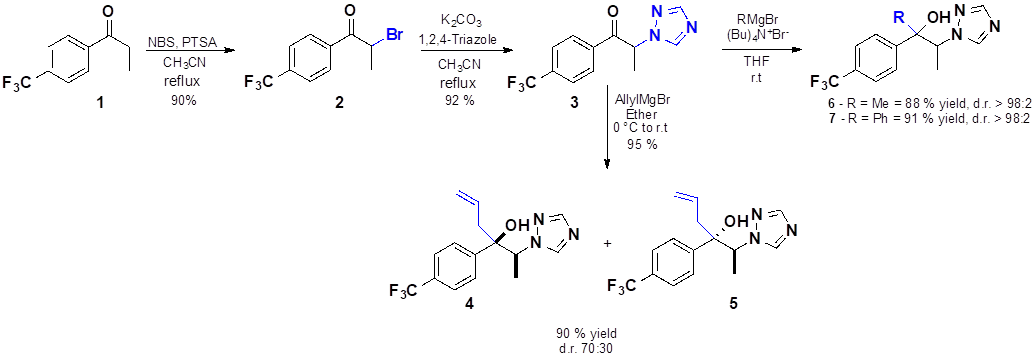
Synthesis of 2-bromo-1-[4-(trifluoromethyl)phenyl]propan-1-one (2). This compound was prepared according to the procedure described in the literature.[12] To a solution of 1.0 g (4.94 mmol) of 4’-(trifluoromethyl)propiophenone 1, (1.41 g, 7.41 mmol), PTSAH2O and NBS (0.88 g, 4.94 mmol) in 25 mL of acetonitrile were stirred and refluxed during 5 hours. The solvent was evaporated and the residue was dissolved in water and extracted with ethyl acetate (x3). Organic extracts were dried with Na2SO4, filtered, and evaporated under vacuum. Following purification by flash chromatography (AcOEt:Hexane, 5:95) product 2 was obtained as a colorless oil (1.25 g, 90%). The spectroscopic data is in agreement with the reported in the literature.[12] 1H NMR (CDCl3, 500 MHz): δ 1.93 (d, 3H, J = 6.6 Hz), 5.27 (q, 1H, J = 6.6 Hz), 7.76 (d, 2H, J = 8.2 Hz), 8.13 (d, 2H, J = 8.2 Hz).
2-(1H-1,2,4-Triazol-1-yl)-1-[4-(trifluoromethyl)phenyl]propan-1-one (3). A stirred solution of (2) 1.35 g (4.80 mmol) in acetonitrile (25 mL) were added K2CO3 (1.06 g, 7.68 mmol) and 1,2,4-triazole (0.53 g,7.68 mmol). After refluxing the reaction mixture during 5 h the acetonitrile was removed in vacuum, the residue was suspended in water and extracted with AcOEt, the organic layer was filtered over Na2SO4 and evaporated under vacuum. Purification by flash chromatography (AcOEt:hexane, 70:30) furnished the compound 3 as a pale yellow oil (1.18 g, 92%). 1 H NMR (CDCl3, 500 MHz): δ 1.87 (d, 3H, J = 7.3 Hz), 6.17 (q, 1H, J = 7.3 Hz), 7.79 (d, 2H, J = 8.2 Hz), 7.98 (s, 1H), 8.10 (d, 2H, J = 8.2 Hz), 8.37 (s, 1H). 13C NMR (125 MHz, CDCl3): δ 18.01 (CH3), 59.76 (CH), 121.44 (q, CH. J = 3.87), 123.38 (q, 272.5), 126.19 (q , J=4.1), 129.09 (CH), 135.49 (q, J= 33.65), 136.62 (C), 142.67 (CH), 151.45 (CH), 193.68 (CO). HRMS (ESI): m/z for C12H11F3N3O+ [M + H]+ calculated 270.0849, found 270.0855.
Diastereoisomeric mixture of 2-(1H-1,2,4-triazol-1-yl)-3-(4-(trifluoromethyl)phenyl)hex-5-en-3-ol (4 and 5). Magnesium turnings (1.35 g, 55.54 mmol) and a few small iodine crystal were suspended in anhydrous diethyl ether (10 mL), then 0.075 mL of allyl bromide were added in order to induce the reaction, the reaction was cooled to 0 °C and a solution of allyl bromide 1.43 mL (2.10 g, 17.36 mmol) in diethyl ether (20 mL) was added slowly. The reaction was stirred at room temperature for 3 h and the final concentration of the Grignard reagent was ~0.58 M. To a solution of 3 (0.82 g, 3.20 mmol) in diethyl ether (30 mL) was added a freshly prepared allylmagnesium bromide (1.86 g, 22 mL, 12.81 mmol) at 0 °C. After stirred for 8 h at room temperature, the reaction was quenched by addition of aqueous saturated ammonium chloride and extracted with AcOEt (x2). The organic layers were washed with aqueous statured solution of NaCl, dried over Na2SO4 and evaporated under vacuum. Flash chromatography purification furnished the mixture of diastereoisomers 4 and 5 (70:30) as a colorless oil (0.85 g, 90 %). The diastereoisomeric ratio was determined by 1H NMR from the crude of the reaction.
The diastereoisomeric mixture was separated through chromatography in order to use each diastereoisomer in the bioassay and to obtain the spectroscopic and spectrometric data for each single diastereoisomer:
Less polar diastereoisomer: white solid, m.p. 92 °C. 1H NMR (500 MHz, CDCl3): δ 1.32 (d, 3H, J = 7.0 Hz), 1.92 (dd, 1H, J = 8.15, 14.15 Hz), 2.66 (dt, 1H, J = 6.2, 14.15 Hz), 3.70 (s, 1H, OH), 4.75 (q, 1H, J = 7.0 Hz), 4.91-5.02 (m, 2H), 5.27-5.35 (m, 1H), 7.59 (d, 2H, J = 8.2 Hz), 7.67 (d, 2H, J = 8.2 Hz), 8.04 (s, 1H), 8.24 (s, 1H). 13C NMR (125 MHz, CDCl3): δ 15.65 (CH3), 44.29 (CH2), 63.29 (CH), 77.35 (C), 120.30 (CH2), 125.49 (CHAr), 126.03 (CHAr), 143.33 (CH), 146.19 (C), 151.60 (CH). HRMS (ESI): m/z for C15H17F3N3O [M+H]+ calculated 312.1318, found 312.1320.
More polar diastereoisomer: colorless oil. 1H NMR (500 MHz, CDCl3): δ 1.68 (d, 3H, J = 8.0 Hz), 2.66 (dd, 1H, J = 7.6, 14.0 Hz), 2.78 (dd, 1H, J = 6.7, 14.0 Hz), 4.19 (br, 1H, OH), 4.75 (q, 1H, J = 8.0 Hz), 5.06-5.12 (m, 2H), 5.41-5.49 (m, 1H), 7.35 (d, 2H, J = 8.2 Hz), 7.49 (d, 2H, J = 8.2 Hz), 7.77 (s, 1H), 7.78 (s, 1H). 13C NMR (125 MHz, CDCl3): δ 15.50 (CH3), 42.93 (CH2), 62.84 (CH), 77.99 (C), 120.17 (CH2), 124.14 (q, CF3, J = 271.36 Hz), 125.16 (CH), 125.79 (CH), 129.55 (q, C-CF3, J = 32.4 Hz), 131.68 (CH), 142.97 (CH), 146.88 (C), 151.42 (CH). HRMS (ESI): m/z for C15H17F3N3O [M+H]+ calculated 312.1318, found 312.1324.
3-(1H-1,2,4-Triazol-1-yl)-2-(4-(trifluoromethyl)phenyl)butan-2-ol (6). A solution of compound 3 in anhydrous THF (1.0 M, 3.72 mL) was slowly added in a steady stream to a solution of tetrabutylammonium bromide (0.12 g,0.37 mmol) and a 3.0 M solution in THF of methylmagnesium bromide (1.33 g, 3.72 mL, 11.16 mmol) at 0 °C. After warmed up to room temperature and stirred during 3h, the reaction was quenched by addition of saturated aqueous ammonium chloride (5mL) and extracted with AcOEt (x2). The organic extracts were washed with saturated aqueous NaCl, dried over Na2SO4 and evaporated. Purification by flash chromatography furnished 6 (single diastereoisomer) as a white solid (0.88 g, 88%). m.p. 129 ˚C 1H RMN (CDCl3, 500 MHz): δ 1.30 (s, 3H), 1.32 (d, 3H, J = 6.9 Hz), 4.10 (br, 1H, OH), 4.64 (q, 1H, J = 6.9 Hz), 7.63 - 7.68 (m, 4H), 8.06 (s, 1H), 8.20 (s, 1H). 13C NMR (CDCl3, 125 MHz): δ 15.55 (CH3), 28.15 (CH3), 63.95 (CH), 75.89 (C), 124.21 (q, CF3, J = 270.11 Hz), 125.45 (CH), 129.63 (q, C-CF3, J = 30.32 Hz), 143.38 (CH), 148.05 (C) 151.95 (CH). HRMS (ESI): m/z for C13H15F3N3O+ [M+H]+ calculated 286.1162, found 286.1169.
1-Phenyl-2-(1H-1,2,4-triazol-1-yl)-1-[4-(trifluoromethyl)phenyl]propan-1-ol (7). Magnesium turnings (0.13 g, 5.57 mmol) and a few small iodine crystals were suspended in 6 mL of THF, then bromobenzene (0.59 mL, 5.57 mmol) was added. After stirring the mixture until the magnesium was dissolved (approximately 2h), a solution of the ketone 3 0.5 g (1.86 mmol) in THF (2mL) was added dropwise. Then, the resulting mixture was stirred during 8 h and quenched by the addition of 1 mL of a saturated solution of ammonium chloride, and extracted with 20 mL of AcOEt. The organic extract was separated and washed with a saturated aqueous solution of NaCl, dried over Na2SO4 and evaporated. Purification by flash chromatography furnished 7 (single diastereoisomer) as a slightly yellow oily liquid (0.58 g, 91%). 1H NMR (500 MHz, CDCl3): δ 1.51 (d, 3H, J = 6.75 Hz), 5.45-5.49 (m, 2H), 7.08-7.12 (m, 1H), 7.71-7.20 (m, 1H), 7.38-7.40 (m, 1H), 7.61 (d, 2H, J = 10 Hz), 7.75 (d, 2H, 8.2), 7.80 (s, 1H), 7.99 (s, 1H). 13C NMR (125 MHz, CDCl3): δ 9.56 (CH3), 61.25 (CH), 79.59 (C), 123.78 (q, CF3, J = 267.93), 124.67 (CH), 125.49 (q, CHAr, J = 3.56), 125.87 (CHAr), 127.42 (CH), 128.51 (CH), 129.39 (q, C-CF3, J = 31.75), 143.58 (CH), 144.10 (C), 147.20 (C), 151.74 (CH). HRMS (ESI): m/z for C18H17F3N3O [M+H] + calculated 348.1318, found 348.1324.
2-Methyl-1-(4-(trifluoromethyl)phenyl)propan-1-one (9). This product was prepared following the procedure reported in the literature.[13] Magnesium turnings (0.85 g, 35.06 mmol) and a few crystals of iodine, were suspended in anhydrous THF (15 mL), then 2-bromopropane (4.31 g, 3.29 mL, 35.06 mmol) was added slowly at 0 °C to obtain a fresh solution of Grignard reagent. In another flask 4-trifluoromethylbenzonitrile 8 (2.0 g, 11.68 mmol) and CuBr (I) 23 mg were dissolved in anhydrous THF (23 mL), next the fresh Grignard reagent solution prepared previously was added at 0 °C and the mixture reaction was refluxed for 4 h under inert atmosphere, after which the reaction turned to black color. The quench was done by addition of water (15 mL) at 0 °C; once that the vigorous reaction finished H2SO4 1N (50 mL) was added and the reaction was refluxed during 1 h. Then the solution was basified by addition of NaOH 2N (pH 9-10) and the aqueous layer was extracted with AcOEt (x3), the organic layers were dried with Na2SO4, filtered and evaporated in vacuum. Flash chromatography (hexane:CH2Cl2, 80:20) gave a slightly yellow oil (1.71 g, 68%). 1H and 13C data are in agreement with the values reported in the literature.[13] 1H NMR (500 MHz, CDCl3): δ 1.25 (d, 6H, J = 6.85 Hz), 3.56 (sept, 1H, J = 6.85 Hz), 7.75 (d, 2H, J = 8.6 Hz), 8.06 (d, 2H, J = 8.6 Hz). 13C NMR (125, CDCl3): δ 18.93 ((CH3)2), 35.82 (CH), 123.76 (q, CF3, J = 270.1 Hz), 125.70 (CH), 128.70 (CH), 134.13 (q, C-CF3, J = 35.92 Hz), 138.92 (C), 203.51 (CO).
2-Bromo-2-methyl-1-(4´-(trifluoromethyl)phenyl)propan-1-one (10). This compound was prepared according to the procedure described in the literature [13] as follow: To a mixture of 9 (1.55 g, 7.16 mmol) in AcOEt (23 mL) were successively added DMSO 0.69 g (0.61 mL 8.5 mmol) and 48 % aqueous hydrobromic acid (0.69 g, 0.47 mL, 8.5 mmol) and the reaction was stirred 5 h at 60 °C. The volatiles were evaporated and the crude mixture was purified by flash chromatography (hexane:AcOEt, 95:5) obtaining a colorless oil (1.88 g, 90%). The spectroscopic data is in agreement with the reported in the literature. [13] 1H NMR (500 MHz, CDCl3): δ 2.0 (s, 6H), 7.71 (d, 2H, J = 8.25 Hz), 8.23 (d, 2H, J = 8.25 Hz).
2-Methyl-2-(1H-1,2,4-triazol-1-yl)-1-(4-(trifluoromethyl)phenyl)propan-1-one (11a). This compound was synthetized following the same procedure for the preparation of 3. In this way, the compound 10 (1.88 g, 6.37 mmol), 1,2,4-imidazole (0.70 g, 10.19 mmol) and K2CO3 (1.70 g, 10.19 mmol) in acetonitrile (23 mL) reacted. Purification by flash chromatography (Hexane: AcOEt, 50:50) furnished the pure regioisomers 11a and 11b. The regioisomeric ratio of 11a and 11b (60:40) was determined by 1H NMR from the crude reaction.
Regioisomer 11a. White solid, m.p. 88 °C. 1H NMR (500 MHz, CDCl3): δ 1.97 (s, 6H), 7.40 (d, 2H, J = 8.25 Hz), 7.57 (d, 2H, J = 8.25 Hz), 7.97 (s, 1H,), 8.28 (s, 8.28). 13C NMR (125 MHz, CDCl3): δ 26.18 (CH3), 68.00 (C), 123.29 (q, CF3, J = 272.85 Hz), 125.59 (CH), 128.50 (CH), 133.95 (q, C-CF3, J = 33.15 Hz), 141.22 (CH), 152.17 (CH), 196.75 (CO). HRMS (ESI): m/z for C13H13F3N3O+ [M+H]+ calculated 284.1005, found 284.1003.
Regioisomer 11b. Colorless oil. 1H NMR (500 MHz, CDCl3): δ 1.24 (s, 3H), 1.26 (s, 3H), 7.65-7.67 (m, 2H), 7.79-7.81 (m, 2H), 8.0 (s, 1H), 8.33 (s, 1H). 13C NMR (125 MHz, CDCl3): δ 19.83 (CH3), 20.11 (CH3), 68.56 (C), 77.13 (C), 123.69 (q, CF3, J = 271.6 Hz), 125.49 (q, CH, J = 4.2 Hz), 127.60 (CH), 131.5 (q, J = 3.38 Hz, C-CF3), 137.61 (C), 142.55 (CH), 152.37 (CH), 205 (CO).
3-Methyl-3-(1H-1,2,4-triazol-1-yl)-2-(4-(trifluoromethyl)phenyl)butan-2-ol (12). The title compound was synthetized following the methodology previously described for (7) using 11a (0.45 g, 1.58 mmol), tetrabutylammonium bromide (51 mg, 0.158 mmol) and methyl magnesium bromide (1.58 g, 3 M, 4.76 mmol) in anhydrous THF (15 mL) at r.t. After flash chromatography purification (AcOEt: hexane, 50:50) furnished a colorless oil (0.49 g, 94 %). 1H NMR (500 MHz, CDCl3): δ 1.45 (s, 3H), 1.59 (s, 3H), 1.68 (s, 3H), 5.0 (br, 1H, OH), 7.46 (d, 2H, J = 8.25 Hz), 7.57 (d, 2H, J = 8.25 Hz), 8.02 (s, 1H), 8.06 (s, 1H). 13C NMR (125 MHz, CDCl3): δ 23.46 (CH3), 23.79 (CH3), 24.32 (CH3), 66.73 (C), 78.56 (C), 124.0 (q, CF3, J = 270.33 Hz), 124.59 (CH), 127.32 (CH), 129.67 (q, C-CF3, J = 31.83 Hz), 142.03 (CH), 147.15 (C), 151.65 (CH). HRMS (ESI): m/z for C14H17F3N3O+ [M+H]+ calculated 300.1318, found 300.1319.
2-Methyl-1-phenyl-2-(1H-1,2,4-triazol-1-yl)-1-(4-(trifluoromethyl)phenyl)propan-1-ol (13). The title compound was synthetized following the methodology previously described for (7) using (0.60 g, 1.66 mmol) of 11a, phenylmagnesium bromide (0.90 g, 4.98 mmol) in anhydrous THF (15 mL). The flash chromatography purification (AcOEt: hexane, 50:50) furnished a colorless oil (0.69 g, 90%). 1H NMR (500 MHz, CDCl3): δ 1.79 (s, 3H), 1.81 (s, 3H), 6.25 (s, OH), 7.22-7.24 (m, 3H), 7.30-7.32 (m, 2H), 7.46 - 7.50 (m, 4H), 7.86 (s, 1H), 8.31 (s, 1H). 13C NMR (125 MHz, CDCl3): δ 23.34 (CH3), 67.34 (C), 82.43 (C), 124.03 (q, CF3, J = 271.93 Hz), 124.43 (q, CH, J = 4.17 Hz), 127.49 (CH), 127.71 (CH), 127.99 (CH), 128.54 (CH), 129.30 (q, CF3 -C, J = 30.95 Hz), 141.70 (CH), 143.56 (C), 148.42 (C), 151.37 (CH).
2-Methyl-2-(1H-1,2,4-triazol-1-yl)-3-(4-(trifluoromethyl)phenyl)hex-5-en-3-ol (14). The title compound was synthetized by a method similar to that for (7) using 11a (0.5 g, 1.76 mmol), allylmagnesium bromide (0.58 M in diethyl ether solution) (1.02 g, 12.2 mL, 7.06 mmol). Flash chromatography purification furnished a slightly yellow oil (0.54 g, 95 %). 1H NMR (500 MHz, CDCl3): δ 1.61 (s, 3H), 1.69 (s, 3H), 2.11 (dd, 1H, J = 8.1, 14 Hz), 3.11 (ddt, 1H, J = 1.25, 5.85, 14.3 Hz), 4.09 (br, 1H, OH), 5.01-5.08 (m, 2H), 5.28-5.38 (m, 1H), 7.41 (d, 2H, J = 7.65 Hz), 7.57 (d, 2H, J = 7.65 Hz), 8.01 (s, 1H), 8.06 (s, 1H). 13C NMR (125 Hz, CDCl3): δ 23.58 (CH3), 23.72 (CH3), 39.85 (CH2), 66.53 (C), 79.54 (C), 120.25 (CH2), 124.40 (q, CF3, J = 272.38 Hz), 124.64 (CH), 128.06 (CH), 129.65 (q, C-CF3, J = 31.42 Hz), 132.53 (CH), 142.66 (CH), 144.86 (C), 151.19 (CH). HRMS (ESI): m/z for C16H19F3N3O+ [M+H]+ calculated 326.1475, found 326.1477.
1-Phenyl-2-(1H-1,2,4-triazol-1-yl)-1-[4-(trifluoromethyl)phenyl]propyl acetate (16). Compound 7 (0.1 g, 0.28 mmol), DMAP (3.42 mg, 0.028 mmol), and DIPEA (0.030 mL 0.022 g, 0.63 mmol) were dissolved in 4 mL of DCM. After stirring the mixture for 10 min 0.022 mL of acetyl chloride (24 mg, 0.30 mmol) was added dropwise. Then, after stirring the resulting mixture during 12 h it was diluted with 15 mL of DCM and washed with a mixture of a saturated aqueous solution of NaHCO3 (5 mL), and a saturated aqueous solution of NaCl (5 mL). The organic extracts were separated, dried over Na2SO4, filtered and evaporated under vacuum. Purification by flash chromatography furnished 16 as a colorless oil (80 mg, 72 %). 1H NMR (500 MHz, CDCl3): δ 1.48 (d, 3H, J = 6.65 Hz), 2.67 (s, 3H), 6.68 (q, 1H, J = 6.65 Hz), 7.06-7.09 (m, 1H), 7.13-7.16 (m, 2H), 7.36-7.38 (m, 2H), 7.62 (d, 2H, J = 8.4 Hz), 7.80-7.81 (m, 3H). 13C NMR (125 MHz, CDCl3): δ 16.04 (CH3), 20.89 (CH3), 60.76 (CH), 79.90 (C), 122.07 (q, CF3, J = 269.43), 125.05 (CH), 125.48 (q, CH, J = 3.47), 125.92 (CH), 127.32 (CH), 128.34 (CH), 129.39 (q, C-CF3, J = 33.28), 141.60 (C), 144.33 (C), 150.45 (CH), 168.19 (CO). HRMS (ESI): m/z for C20H19F3N3O2 [M+H]+ calculated 390.1429, found 390.1426.
tert-Butyl [1-phenyl-2-(1H-1,2,4-triazol-1-yl)-1-(4-[trifluoromethyl]phenyl)propyl] carbonate (17). Di-tert-butyldicarbonate (64 mg, 0.28 mmol) and DMAP (1.7 mg, 0.014 mmol) were added to a solution of 7 (0.05g, 0.14 mmol) in 4 mL of CH3CN and the resulting solution was stirred for 12 h at room temperature. Then, the mixture reaction was diluted with AcOEt (15 mL) and washed with both a saturated aqueous solution of NaCl (5mL) and a saturated aqueous solution of NaHCO3 (5mL). The organic extract was dried over Na2SO4, filtered and evaporated under vacuum. Purification by flash chromatography furnished 17 as a colorless oil (20 mg, 32 %). 1H NMR (500 MHz, CDCl3): δ 1.35 (s, 9H), 1.60 (s, 3H), 6.29 (q, 1H, J = 7.15 Hz), 7.28-7.29 (m, 3H), 7.36-7.38 (m, 4H), 7.57 (d, 2H, J = 8.3 Hz), 7.64 (s, 1H), 7.84 (s, 1H). 13C NMR (125 MHz, CDCl3): δ 17.30 (CH3), 27.59 [(CH3)3], 58.57 (CH), 83.0 (C), 87.60 (C), 124.29 (C), 128.08 (CH), 128.37 (C), 128.78 (CH), 129.24 (CH), 150.59 (CO). HRMS (ESI): m/z for C23H25F3N3O [M+H]+ calculated 448.1843, found 448.1848.
2-(1H-1,2,4-Triazol-1-yl)-1-(4-(trifluoromethyl)phenyl)propan-1-ol (19): To a solution of ketone 3 (0.5 g, 1.85 mmol) in MeOH (10 mL) at 0 °C NaBH4 (0.21 g, 5.57 mmol) was added and the resulting mixture was stirred at room temperature during 2 h. Saturated aqueous solution of NH4Cl (1mL) was added. After evaporation, the residue was suspended in water (15 mL) and extracted with 15 mL of AcOEt (x2), the organic extracts were dried over Na2SO4, filtered and evaporated. After flash chromatography (AcOEt: hexane, 70:30) both diastereosiomers were isolated as a white solid (0.4 g, 80%). The diastereoisomeric ratio (60:40) was determined by 1H NMR from the crude reaction. A second flash chromatography (AcOEt:hexane, 60:40) allows separate each single diastereoisomer.
Less polar diastereoisomer: white solid, m.p. 97 °C. 1H NMR (500 MHz, CDCl3): δ 1.45 (d, 3H, J = 7.0 Hz), 4.61 (qd, 1H, J = 3.6, 7.0 Hz), 4.78 (br, 1H, OH), 5.16 (d, J =3.6 Hz), 7.44-7.46 (m, 2H), 7.61 (d, 2H, J = 8.1 Hz), 7.85 (s, 1H), 7.98 (s, 1H). 13C NMR (125 MHz, CDCl3): 13.22 (CH3), 61.44 (CH), 74.44 (CH), 124.13 (q, J = 270.77, CF3), 125.46 (q, J = 3.6, CH), 126.35 (CH), 130.20 (q, J = 32.43, C-CF3), 142.16 (CH), 144.05 (C), 151.30 (CH). HRMS (ESI): m/z for C12H13F3N3O [M+H]+ calculated 272.1005, found 272.1010.
More polar diastereoisomer: white solid, m.p. 108 °C. 1H NMR (500 MHz, CDCl3): δ 1.47 (d, 3H, J = 7.0 Hz), 4.64 (br, 1H, OH), 4.52 (qd, 1H, J = 6.9 Hz), 4.96 (d, 1H, J = 6.6 Hz), 7.35 (d, 2H, J = 8.5 Hz), 7.59 (d, 2H, J = 8.0 Hz), 7.87 (s, 1H), 7.95 (s, 1H). 13C NMR (125 MHz, CDCl3): 17.08 (CH3), 61.63 (CH), 75.98 (CH), 124.05 (q, J = 270.0, CF3), 125.60 (q, J = 4.06, CH), 126.70 (CH), 130.52 (q, J = 32.45, C-CF3), 143.19 (CH), 144.44 (C), 151.63 (CH). HRMS (ESI): m/z for C12H13F3N3O [M+H]+ calculated 272.1005, found 272.1011.
Bioassays. First, it was selected Fusarium solani as a model since this fungus is also pathogenic to 111 tree species, including the avocado trees.[14] The antifungal activities of the title compounds (3 to 7, 11 to 14, 16 and 17) and the commercial fungicide propiconazole were evaluated and compared according to the next procedure: 4 µL of conidia suspension of Fusarium solani strain were inoculated on each well of a 12-well cell culture plates with 1 ml of solid PDA (potato-dextrose-agar) culture media supplemented with 1 mM of the propiconazole derivatives (3 to 7, 11 to 14, 16 and 17). The plates were incubated at 29±1°C in complete darkness for 5 days. Solid PDA culture media and solid PDA culture media supplemented with methanol 1mM were used as controls by duplicate. Solid PDA culture media supplemented with propiconazole 1 mM was used as a positive control. The antifungal activity was recorded at 3, 4 and 5 days after inoculation (dpi) with a camera (Nikon model 3200) and the area of fungus growth including controls were obtained with image processing software (ImageJ). Since we did not observe a significant difference in the area with the methanol control, only the control with PDA was considered to calculate the percentage of inhibition. At 4 dpi the antifungal activity of the propiconazole derivatives were evaluated and the percentage of growth and percent inhibition of the growth of the fungus was calculated with the formula: % inhibition = 100-(Area of the fungus exposed to the compound * 100) / Control area. Finally, the most relevant compounds were tested against the Fusarium euwallaceae strain HFEW-16-IV-019 that was provided by the Mycology Laboratory at CNRF (Centro de Referencia Fitosanitaria) of SAGARPA, Mexico’s agriculture ministry.
The phytotoxicity evaluation of all compounds was also studied and compared according to the next procedure: Arabidopsis thaliana seeds were disinfected with 96% ethanol during 5 min and then with sodium hypochlorite for another 5 min. Next, the seeds were rinsed with sterile distilled water and stored at 4 ˚C for 48 hours for late use. The disinfected seeds were germinated in MS 0.2x medium (Murashige and Skoog, Sigma Aldrich, Cat. M5524), and supplemented with each novel compounds. Finally, the Petri dishes that contain the seeds and the novel compound were incubated in the plant growth chamber with a photoperiod of 16 hours lights and 8 h darkness at 22 ˚C with a relative humidity of 80%. The analysis of root growth was performed after 10 days of germination. The length of the primary root, number and density of lateral roots were measured with a simple ruler. The number of lateral roots was determined by counting the lateral roots present in the primary root. The lateral root density was determined by dividing the number of lateral roots between the length of the primary root and expressed as NRL/cm.
Results and Discussion
Synthesis of all compounds. The synthetic route of compounds 2 to 7 is outlined in Fig. 3. Compound 2 was prepared via α-halogenation in 90% yield starting from 4-trifluoropropiophenone 1 and N-bromosuccinimide. Nucleophilic substitution of compound 2 with 1,2,4-triazole gave 3 in 92 % yield. Tertiary alcohols 6 and 7 were prepared from ketone 3 in 88 and 91 % yield, respectively, by addition of Grignard reagent in the presence of tetrabutylammonium bromide. The Felkin-Anh model suggests that this reaction should proceed with high selectivity, and the experimental result is in agreement with it, since the product was obtained with a diastereoselectivity up to 98%. In order to prepare compounds 4 and 5 it was used allyl magnesium bromide as the Grignard reagent and the compound 3. The diastereoisomeric mixture of compounds 4 and 5 (d.r. = 70:30) was separated through chromatography to obtain a sample of each single diastereoisomers. All the compounds were stable at room temperature for several days.
The target compounds 9 to 14 were synthesized from 4-trifluoromethylbenzonitrile 8 as show in Fig. 4. Benzonitrile reacted with the Grignard reagent prepared “in situ” from 2-propylbromide and magnesium, in the presence of iodine and copper bromide (I) to obtain the ketone 9 in 68 % yield. Compound 10 was prepared via α-halogenation with HBr/DMSO in 90% yield. Nucleophilic substitution of compound 10 with 1,2,4-triazole gave a regioisomeric mixture of 11a and 11b (ratio 60:40) in 54 and 30 % yield, respectively, this result is explained based on the annular tautomerism of 1,2,4-triazole.[15]. The triazol 11a was separated by crystallization and reacted with the appropriate Grignard reagent to prepare the target compounds 12, 13 and 14. For example: MeMgBr gave 94 % yield, while PhMgBr and allylMgBr, furnished the desired product in 90 and 95 % yield, respectively.

As show in Fig. 5, the alcohol 14 was reacted with di-tert-butylcarbonate, DMAP, and DIPEA in dichloromethane to obtain the product 15 in 22 % yield. The acetylation of tertiary alcohol 7 with acetyl chloride and DMAP proceeded in 78% yield to give 16. On the other hand, alcohol 7 was also reacted with di-tert-butylcarbonate, and DMAP to furnished 17 in 33% yield. All this reaction gave the O-protected tertiary alcohols.

Finally, secondary alcohol 19 was obtained through the reduction of ketone 3 with sodium borohydride in methanol at 0 ˚C to r.t., to furnish 19 as a mixture of diastereomers (r.d. 60:40) (Fig. 6).

The diastereoisomeric mixture was separated by chromatography and each single diastereoisomer was analyzed by NMR, HRMS-QTOF and used in the bioassays.
Structure-Activity Relationship. The antifungal activities for compounds 3 to 7, 11 to 14 and 16 and the commercial fungicide propiconazol were tested, and the results of inhibition rates (%) are summarized in Fig. 7. The 1H-1,2,4-triazole compounds’ mode of action is the arrest of sterol biosynthesis by inhibiting 14α-desmethylase, which is a specific cytochrome P450. Evidence that sterol biosynthesis inhibitions is linked to the binding of nucleophilic N4 of 1,2,4-triazole to iron in the ferric state of the heme is an essential feature of the inhibition action.[16] On the other hand, the van der Waals radius of fluoride is 1.47 Å[16] which is only 20% larger than that of hydrogen and much more smaller than those of other halogens. The C-F bond length in CH3F is 1.382 Å, which is 0.295 Å longer than the C-H bond in methane, but 0.403 and 0.551 Å shorter than the C-Cl and C-Br bonds, respectively. Because of this similarity in size to hydrogen, it has been shown that microorganism or enzymes often do not recognize the difference between a natural substrate and its analogue wherein a C-H bond of the substrate is replaced with a C-F bond. Furthermore, since the H3C-F bond is stronger than that of H3C-H by 5.0 kcal/mol, the replacement of a specific C-H bond with a C-F bond can effectively block metabolic processes via hydroxylation of C-H bonds, predominantly by the cytochrome P-450 family enzimes. It means, strategic incorporation of fluorinated groups into the metabolism site could prevent deactivation of biologically active substances in vivo.[17] As shown in Fig. 7, most of the title compounds had a good inhibition rate at 1mM concentration; in all cases, when the hydroxyl group is protected with Boc (compounds 17) or Ac (compound 16) protecting groups, the biological activity is diminished. It means, that both the hydroxyl group and the 1,2,4-triazole are indispensable to inhibited the oxidative removal of the sterol C(14) groups by the cytochrome P450 enzyme, which is necessary in order to have antifungal activity.

It was also studied the diastereoisomeric mixture and each single diastereoisomer (compounds 4 and 5). The mixture of syn and anti compounds were less active, and the more polar diastereoisomer 4 shows better antifungal activity when compared to less polar diastereoisomer 5, with inhibition rates of 50 and 17%, respectively. When it is compared compounds 6 and 7, it was observed that the substituents in R affect dramatically the antifungal activity, for instance, when R is a methyl group the inhibition ratio is 10% while R is phenyl the inhibition ratio is higher than 80%. The ketone 3 was also evaluated and shows up to 48% inhibitory activity.
Next, the compounds 11, 12, 13, and 14 which contain the 1,2,4-triazole-α,α-dimethyl moieties were also bioassayed and it was found 32, 32, 65, and 31 % inhibitory activity, respectively.
The secondary alcohols 19, 19A and 19B were also prepared; the lack of an extra substituent on C-1 has a negative effect in the biological activity as fungicide. It was shows a percent inhibitory activity of 4.8, 7.3, and 12.1%, respectively. Also, it was bioassayed the mixture of compounds 7 and 3 because of the good fungicide activity and low phytotoxicity. Unfortunately, the inhibitory activity against Fusarium solani was less than 10%.
Finally, in order to increase the scope of this research the compounds 7 and 13 were selected because of their good inhibitory activity and low phytotoxicity and tested against the extremely pathogenic fungus Fusarium euwallaceae at 100 (M (table 2). The newly synthesized triazole derivatives 7 and 13 exhibited good antifungal activity since both inhibited the growth of the fungus up to 63 % (Fig. 8).

Phytotoxicity evaluation of target compounds. The analysis of the phytoxicity for compounds 3 to 5, 7, 11 to 14, 16 and 17, and the commercial fungicide propiconazole are summarized in Fig. 9. The results revealed that in most cases the length of the primary root was higher indicating a less phytotoxic effect when compared to commercially available propiconazole. Compound 7 had a negative effect in the length of the primary root by 14.77% (see PCZ vs 7, in graph 3), which indicates a phytotoxic effect. On the other hand, the ketone 3 shows a very interesting lack of phytotoxicity among all other compounds, this result suggest that this compound could have a promising applicability as fungicide.

In conclusion, a series of new 1,2,4-triazole derivatives bearing a p-trifluoromethylphenyl moiety were synthesized and evaluated for antifungal activity in vitro against Fusarium solani, and the extremely phytopathogenic fungus Fusarium euwallaceae; also the phytotoxic effect was evaluated using Arabidopsis thaliana. Almost all the compounds showed from modest to good growth inhibition activity of the fungi at 1 mM and even at 100 (M when tested against Fusarium euwallaceae. The compounds 7, 13, and 4 showed great potential to be developed as new antifungal agents because its good antifungal activity and also because of their low phytotoxicity. SAR showed that free alcohols and not O-protected compounds significantly influence the activity. Hence, (-methyl-(-1,2,4-triazole emerged as novel compound to develop new ketone-triazole-type antifungal agents. Further research on structural modification and evaluation as fungicides is ongoing.

