











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Arsenic is an element which is associated with a wide range of adverse health effects and is extensively distributed through the earth crust [1-2]. Nevertheless, arsenic presence on drinkable water is usually associated with natural earth deposits, arsenic contamination may also occur as a consequence of anthropogenic activities such as: manufacturing metals and alloys, refining petroleum, and burning fossil fuels and wastes [2]. These circumstances have created a strong legacy of arsenic worldwide pollution through exposure to inorganic arsenic via drinking water turning it into a growing public health concern [3]. Currently, contamination of groundwater with As is one of the most important health risks as source of drinking and irrigation water supplies [4]. In this sense, toxic and carcinogenic properties of As inorganic species make their quantification in natural water actually important [5].
Inorganic arsenic exists in groundwater mainly in two chemical forms. The most abundant (80% ca.) is arsenite (H3AsO3) which is 10-50 times more toxic than the arsenate ions (H2AsO4 - or HAsO4 2-) owing their reaction with enzymes in the human respiratory system [2,6]The maximum arsenic concentration allowed in drinking water (Mexico legal limit) is 25 μg L-1 [7] but a recommended level of 10 μg L-1 is set by the World Health Organization (WHO) [8].
A variety of well-known and accurate analytical techniques are capable of measuring the arsenic concentrations in water samples, as Atomic Absorption Spectroscopy (AAS), Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES), Gas Chromatography (GC), High Performance Liquid Chromatography (HPLC) or Capillary Electrophoresis (CE), for instance [9-10]. These methods are expensive and laboratory-based demanding well trained technicians to conduct the measurements; thus, are not suitable for field detection [2,10]. Therefore, there is an increasing need to develop portable devices which gives reliable measurements for the detection of arsenic at low cost. Currently, the development of electrochemical methods for arsenic detection is and active research area due to the sensitivity, portability and short time required to perform the measurements [6]. Electroanalytical methods such as stripping voltammetry, potentiometry and differential-pulse polarography have been reported for determination of very low concentrations of arsenic in natural waters samples [4,11-13]. Electroanalytical techniques provide a low cost, rapid and portable option for routine in-field monitoring of large numbers of samples. According to their sensitivity, anodic stripping voltammetry (ASV) has been established as the most important technique for the determination of trace amounts of arsenic. ASV comprises the preconcentration of As (0) produced from reduction of As (III) and deposited on the electrode surface followed by anodic stripping of As (0) [12]. In order to improve this analytical procedure several working electrode have been used as Au, Pt, Ag, Hg, boron doped-diamond (BDD) or nano-particle and carbon nano-tube modified electrodes which reports limits of detection from 0.0026 to 170 μg L-1 [11,13].
Nevertheless, some of these techniques could be turned into a portable arsenic detector, the use of costly materials surfaces would likely make them prohibitively expensive [14], while mercury electrodes have a restricted use in electrochemical analysis [15]. An alternative approach is to support metal nanoparticles onto glassy carbon or nanotubes substrates for As electroanalytical determination [6,16,17]. In the present paper, we proposed a simple and low-cost procedure for the determination of arsenic (III) in water samples by means of square wave anodic stripping voltammetry at screen printed and glassy carbon electrodes by means of co-deposition of arsenic with silver.
Experimental
Chemicals
All solutions were prepared with 18 MΩ cm-1 deionized Milli Q (Millipore) water with chemicals of analytical grade without further purification. As (III) stock solution was prepared from sodium (meta) arsenite NaAsO2 (Fluka, 99%) in 0.1 M nitric acid (Sigma-Aldrich, 70%). Ag (I) stock solution was prepared by dissolving AgNO3 (Fluka, 99.5%) in 0.1 M nitric acid. Solution of As (III) with concentration 10-3 M was prepared daily. KCl (Sigma-Aldrich) was used for chlorine interference study. Elemental solutions (Cu, Hg, Pb) used for interference experiments were prepared from Cu(NO3)2•5H2O (Aldrich), Hg(NO3)2•H2O (Fluka) and Pb(NO3)2 (J.T. Baker) dissolving in HNO3 0.1 M.
Construction of carbon screen printed electrodes
Manual screen printing equipment with a conventional monofilament polyester mask of 62 threads/cm2, was used for the fabrication of the carbon planar electrodes which were printed over a styrene sheet as substrate (thickness: 0.762 mm). Electrodag PF-407C (Acheson) commercial thermoplastic carbon paste, was used as base material for the printed electrodes. After the printing process, a drying cycle of 120 minutes was followed. Once the printed carbon strips were dried, they were partially covered with an insulating layer of Electrodag 451 SS dielectric ultraviolet photocurable ink, allowing a carbon exposed area of 15 mm2. A polyurethane squeegee was used for the printing process.
Instrumentation
Electrochemical measurements were recorded using a PGSTAT 30 AUTOLAB electrochemical system (Ecochemie). The electrochemical cell consisted of a planar carbon screen printed strip or a glassy carbon electrode (BAS MF-2012, area of 7.1 mm2) as a working electrode, a platinum wire electrode (XM110 Radiometer-Copenhagen) as counter electrode and a double junction Ag/AgCl (900200 Orion) with 10% KNO3 external solution as a reference electrode. All measured potentials are referred to this electrode and matches their potential characteristics with of a conventional KCl saturated calomel electrode SCE (E=SCE). The experiments were performed at room temperature using nitric acid 0.1 M as supporting electrolyte. The high peak current is reported with respect to a tangent line fitted to the peak’s base. In the anodic stripping voltammetry experiments, the response is enhanced applying a deposition potential while the solution is stirred. In order to minimize the time of analysis and to simplify the analytic process all experiments were done without deoxygenating the solutions [18]. As(III) was also determined by means of Hydride Generation Atomic Absorption Spectroscopy using an atomic absorption spectrometer SpectrAA 880 (Varian) as a reference for the analysis of water spiked samples.
Results and Discussion
It is well known that As(III) is not electroactive at conventional carbon electrodes; consequently, these electrodes are not suitable for arsenic voltammetric determination. Therefore, gold or mercury films onto carbon electrodes have been recommended, but with cost and environmental significant consequences. In these sense, co-deposition of arsenic and silver can be an attractive alternative for the arsenic quantification.
Figure 1 shows the behavior of an arsenic (III) not electroactive glassy carbon electrode in HNO3 0.1 M. On the other hand, when As(III) is co-deposited with silver, the reduction and oxidation of arsenic can be distinguished clearly at -0.35 and 0.15 V respectively.
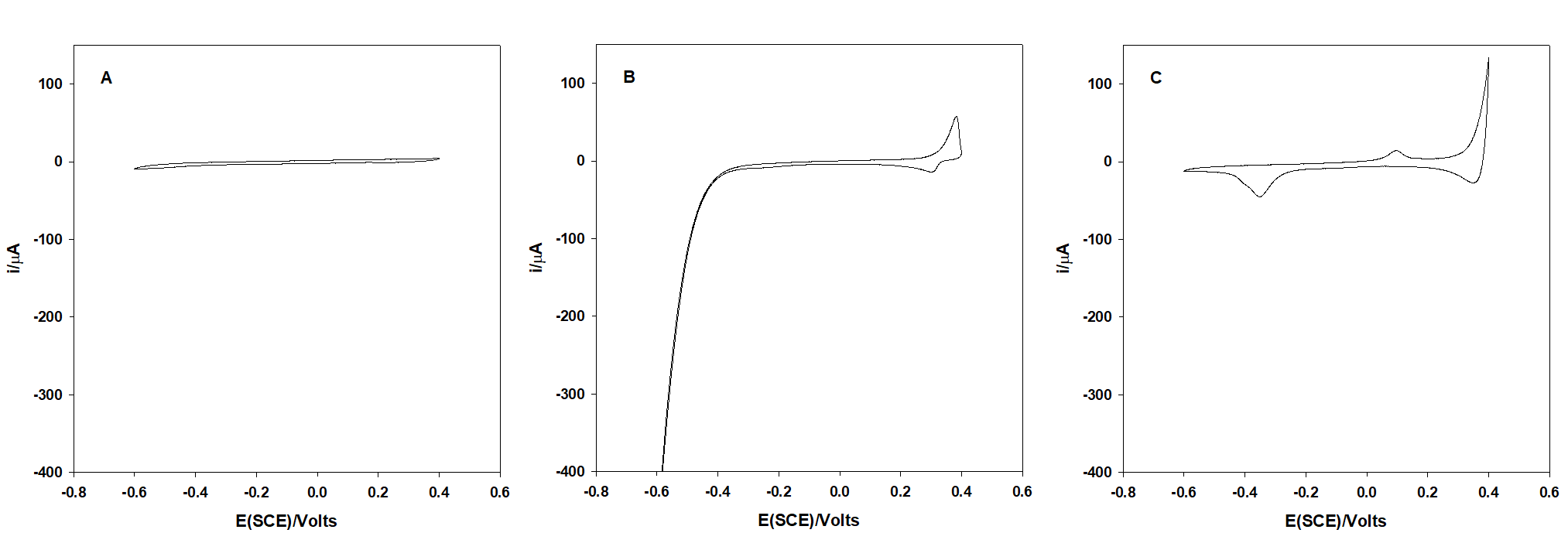
Fig. 1 Cyclic voltammograms in HNO3 0.1 M at a glassy carbon electrode. (A) As(III) 7.5 mg mL-1, (B) Ag+ 15 mg mL-1, (C) As(III) 7.5 mg mL-1 + Ag+ 15 mg mL-1. Conditions: 20 mV s-1 from OCP (0.18 V) toward the cathodic direction for 1A; CVs 1B and 1C were recorded from a constant potential of 0.18 V.
Once established the suitably of the proposed strategy for the arsenic determination, it was obtained the voltammogram of As(III) by means of square wave anodic stripping voltammetry in order to increase the sensitivity and reduce interferences (Fig. 2). The stripping signal obtained, previous deposition of As(III), confirms that is possible to quantify As (III) by co-deposition with silver onto carbon electrodes.
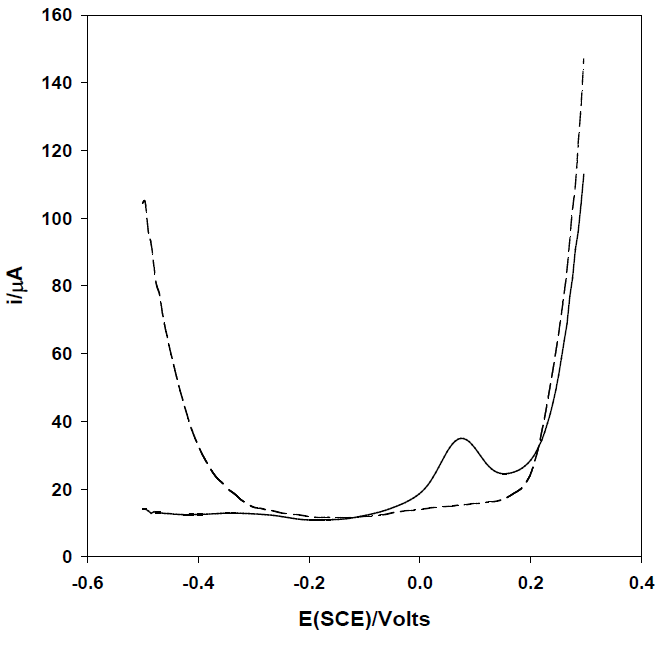
Fig. 2 Square wave anodic stripping voltammogram of As(III) 0.06 mg mL-1 in 0.1M HNO3 M with Ag+ 2 mg mL-1 using a glassy carbon electrode. Conditions: Edep = -0.5 V, tdep = 180 s, A=50mV, s=10 mV, F=100 Hz). Dashed line represents the baseline of the supporting electrolyte.
Optimization
Increasing the intensity of the signal in SWASV is an important optimization stage because this implies the development of analytical methodologies with smaller detection limit and enhanced signal/noise ratio. Therefore in the present work, an optimization methodology was performed.
Chemometric tactic have been frequently applied to the optimization of analytical methods, because resulting in lower reagent consumption and considerably less laboratory work. In this sense, Box-Benhken is an efficient design for response surface modeling since permits the estimation of the parameters of a quadratic model and the percentage of response which is explained by its relationship with the variables [19]. The optimization step was performed using a Box Behnken design for both working electrodes: screen printed and glassy carbon for a 50 µg mL-1 As(III) solution.
In SWASV the most important factors to be optimized are deposition potential (Edep), time of deposition (tdep), frequency (F), amplitude (A) and step potential (S). From preliminary experiments and references Edep = -0.5 V, S = 10 mV, tdep = 180 s were fixed [18]. A, F and silver concentration (C) were selected as independent variables for the Box-Behnken optimization process. The uncoded factor levels used for both working electrodes are reported at Table 1.
Table 1 Uncoded factor levels for the Box-Behnken design BBD
| Factor | - | 0 | + |
|---|---|---|---|
| Silver concentration (C) mg mL-1 | 2 | 6 | 10 |
| Amplitude (A) mV | 10 | 55 | 100 |
| Frequency (F) Hz | 10 | 55 | 100 |
Owing to the three factors and 3 replicates of the central point, it was necessary to perform 15 experiments. All the experiments were carried out in random order and results were processed by means of Minitab 17.
The equation (1) models the intensity of the peak (ip) as function of amplitude, frequency and silver concentration for the glassy carbon electrode with a coefficient of determination of 0.929. This equation predicts a maximum of the peak current of 50.04 µA at A = 100 mV, F = 100 Hz and C = 2 mg mL-1 which are considered the optimal conditions. The experimental value obtained for ip was 49.97 (n = 6; %RSD = 1.83).
In contrast, the equation (2) models the intensity of the peak (ip) as function of amplitude, frequency and silver concentration for the screen printed carbon electrode with a coefficient of determination of 0.883. This equation predict a maximum of the peak current of 31.77 µA at A = 100 mV, F = 100 Hz and C = 10 mg mL-1 which are considered the optimal conditions. The experimental value obtained for ip was 31.05 (n = 6; %RSD = 6.90).
The lower sensitivity of the screen printed electrodes is attributed to its larger resistivity. This effect is partially compensated with a greater concentration of silver as a result of the optimization process.
Analytical parameters
From the calibration curve carried out at the previously selected conditions for the glassy carbon electrode, a linearity range of 10 to 60 µg L-1 (r2 ≥ 0.99) was found, with a sensitivity of 0.98 µA L µg-1. A detection limit of 4.2 µg L-1 (3.3 σ/S) was obtained from the standard deviation of the response (σ) and the slope (S) of calibration curves. Precision was evaluated by measuring the peak height current of 10, 50 and 80 µg L-1 As(III) solutions (sixfold). The percent relative standard deviations found were of 6.0, 4.7 and 3.5% respectively, which is equivalent with the precision usually reported by SWASV.
Calibration curve for the carbon screen printed electrode shows a linearity range of 10 to 80 µg L-1 (r2 ≥ 0.99) with a sensitivity of 0.60 µA L µg-1 with a detection limit of 8.4 µg L-1. Precision was also evaluated by measuring the peak height current of 10, 50 and 80 µg L-1 As(III) (sixfold). The percent relative standard deviations found were of 8.7, 6.3 and 5.9% respectively.
The limit of detection of arsenic obtained with both carbon electrodes is lower than the obtained with a silver electrode (10 μg L-1) [18] which normally has a higher cost, memory effect and a lower voltammetric signal reproducibility due to the formation of an oxide film [13].
Nevertheless carbon screen printing electrodes have greater roughness and physical area than the glassy carbon electrode, the solvents and polymers formulation of the carbon printing inks usually induce low heterogeneous kinetics on the CSPE reducing the co-deposition of As with Ag and therefore their sensitivity.
Quantification of As by means of co-deposition strategy onto carbon electrodes has been also reported using Cu or Bi. Co-deposition of As with bismuth reports a very low limit of detection but a narrow linear range of work [20]; on the other hand co-deposition of As with cooper has been explored but with lower sensitivity and greater limit of detection [21]. In this sense co-deposition of As with Ag is a competitive alternative for the analysis of arsenic.
Interferences
Chloride ion is a chemical species that can be found commonly in real samples. Most of the guidelines for drinking water quality have established a permissible maximum limit of 250 mg/L (7.0 x 10-3 M) [22].
A metallic silver working electrode in acidic media, shows a signal of oxidation located at 0.3 V (SCE); this signal changes to 0,0 V (SCE) in [HCl] = 0.1 M doing impossible the analysis of As (III) [23]. In a silver film deposited onto a carbon working electrode, the presence of chloride also represents a significant interference, because the oxidation signal shift and the silver chloride precipitation which reduces the silver concentration in the system.
To evaluate the effect of chloride on the oxidation peak of [As (III)] = 25 μg L-1 obtained by SWASV, voltammograms on HNO3 0.1M with silver were obtained (Fig. 3). As it was expected, when the concentration of chlorides is increased, the arsenic peak height goes diminishing because the silver concentration is reduced by the precipitation of AgCl and consequently less silver film is deposited onto the carbon electrode. This effect is quite similar with glassy carbon and screen printed electrodes; the height of peak is considerably affected from 3 x 10-4 M and 2 x 10-4 M respectively.
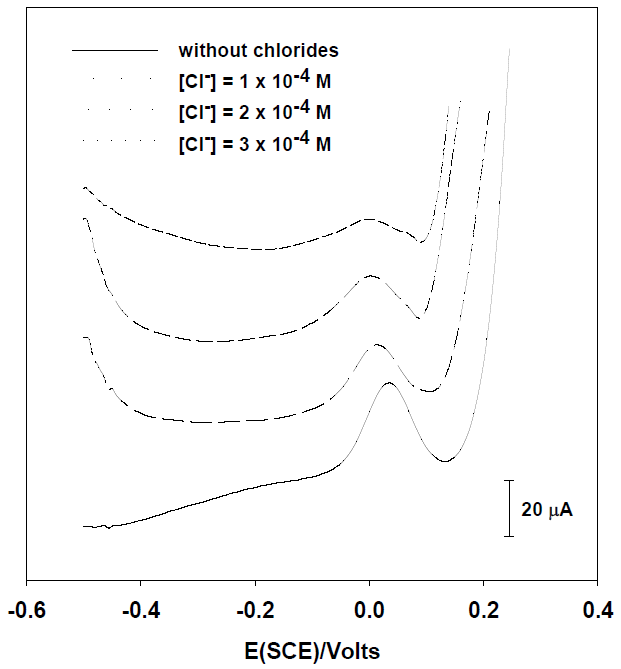
Fig. 3 Square wave voltammograms of [As (III)] = 25 μg L-1 on HNO3 0.1M with 2 mg mL-1 of silver at different chloride concentrations with a glassy carbon electrode.
Mercury, lead and copper ions were evaluated as the heavy metals which seriously interfere in the quantification of arsenic [23,24]. Thus, these metals were successively added to a solution of arsenic 25 μg L-1 in nitric acid 0.1 M to evaluate their effect on the stripping signal of arsenic (Fig. 4). In concordance with previous studies, our results indicate that copper constitutes a significant interferent in the detection of As (III) [18,23,25]. Copper is reduced at more positive potentials than arsenic and it is observed an overlapping of stripping signal of Cu and As which increase the peak height (Fig. 4).
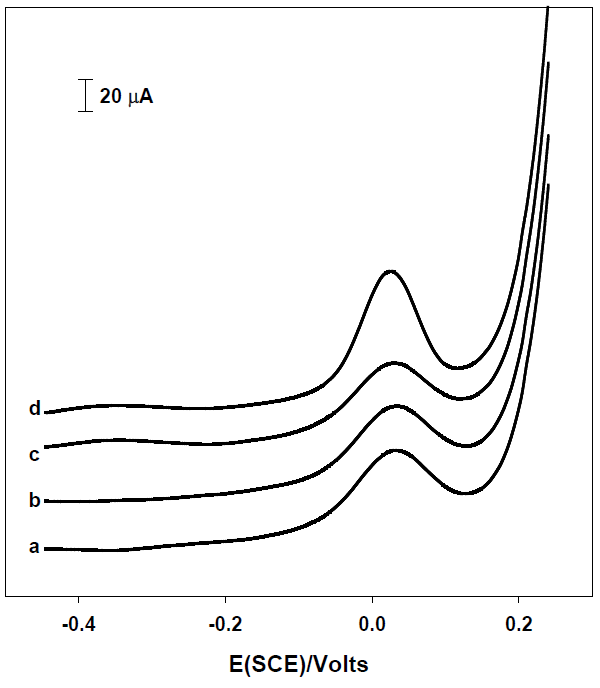
Fig. 4 Square wave voltammograms of [As (III)] = 25 μg L-1 in presence different heavy metal interferences at 25 μg L-1 each one. (a) H3AsO3, (b) a + Hg2+, (c) b +Pb2+, (d) c + Cu2+. Glassy carbon electrode.
The results of this interference study are promising because interferences reported in natural arsenic contaminated groundwater are below the detectable concentration level of interfering ions studied [26].
Application to synthetic water samples
In order to evaluate the utility of the co-deposition of silver in the determination of arsenic, three spiked purified water samples were analyzed by means of SWASV utilizing carbon screen printed electrodes and standard addition methodology. The obtained results were compared with those obtained with hydride generation atomic absorption spectrometry (Table 2) [27].
Table 2 Results of analysis of spiked water samples by SWASV and HG-AAS (mean ± SD n=3)
| SWASV/μg L -1 | AAS/μg L -1 | |
|---|---|---|
| 24.9 ± 2.5 | 25.5 ± 2.2 | |
| 38.9 ± 1.2 | 40.7 ± 1.7 | |
| 75.5 ± 1.9 | 77.0 ± 0.8 |
The concentration of arsenic found by the proposed method and the values obtained using AAS as reference technique, show good agreement and not significant differences were obtained by means of the ANOVA statistical analysis of the results.
Conclusions
This study proposes an easy way to analyze arsenic (III) by means of the co-deposition of silver onto carbon electrodes. The strategy can be implemented in screen printed disposable device which can be used with portable instrumentation for decentralized analysis. The total analysis system can be used for the monitoring of drinking water in places where there is no access to sophisticated instrumentation, but arsenic analysis is required according to the water quality guidelines.
The linearity achieved with the developed methodology includes the permissible limit of arsenic in water. Analytical results demonstrate that the in-situ determination of arsenic in contaminated groundwater of remote regions is feasible by the proposed method and constitute a valuable contribution to resolve a public health concern.

