











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Slowly but inexorably chronic non communicable diseases, such as cardiovascular disease (CVD), diabetes mellitus and chronic kidney disease (CKD), have reached the top as most common causes of premature death[1,2], of these diseases, CKD affects 10% of the population worldwide and around 80% of them occurs in low- or middle-income countries[1]. Notwithstanding a different origin, chronic kidney disease (CKD) is a silent and progressive decline in renal function, which leads to the loss of functional nephrons, and ultimately an end-stage renal disease (ESRD) occurs. Because CKD remains asymptomatic until its late stages, CKD detection is a complex issue, that is most often diagnosed with the onset of kidney failure and there are few chances to effectively prevent adverse outcomes derived from CKD advanced stage[3]. This major health problem defined as reduced kidney function (glomerular filtration rate (GFR) <60 mL/min/1.73m2) and/or evidence of kidney damage (usually albuminuria/proteinuria) for a period of at least three months[4], increases mortality, cardiovascular disease and obviously ESRD risk[5,6]. Despite of this, with early diagnosis and treatment, it's possible to slow or stop the CKD progression, the risk of kidney failure progression and cardiovascular disease can be decreased by up to 50% when early detection occurs, generally through albuminuria/proteinuria assessment[7]. Clinical and experimental evidences indicate that albuminuria and proteinuria (both markers of CKD progression) also have an active role in the disease [5,8].
Although proteinuria it is considered an independent risk factor for cardiovascular (CV) morbidity and mortality, and despite its association with poor renal outcomes, still exists uncertainty with regard to the optimal conditions to its determination, among them, remains unclear if measure total urinary protein, albuminuria or both, as well as the optimum number of urine samples required, a lack of a uniform criteria in regard to whom to test, the existence of non albuminproteinuria (NAP, which reflects tubular damage), aside a lack of standard reference materials, variable definitions and different units included in reports, which concurs in a complex, difficult task yet essential.([5,9,10]
Because of this lack of standardization to this regard, a major concern is the search for reliable analytical methods to effectively assess renal function in early and late stages of CKD, as well as its therapeutic response. This work proposes the determination of two different markers to achieve an adequate evaluation of the renal function in diabetes mellitus type 2 patients. It has been proposed that a reduction in the number of renal epithelial cells or podocytes, may lead to the development of proteinuria [11-13]. Podocyte injury and loss have been observed in human and experimental models of glomerular diseases[14,15], and play a role in the pathogenesis of diabetic nephropathy (DN)([11,16]. Also podocyturia is considered a marker to estimate activity and severity of glomerular damage, because of this, has been used to predict disease progression, being a more sensible and earlier sign of glomerular damage than proteinuria[17]. Nevertheless it is unclear how the histopathological podocyte alterations are involved in the pathogenesis of proteinuria, but several important podocyte molecules have been identified and could serve as the most obvious target for pharmacological intervention in proteinuric diseases[18]. Yet it is unclear if damage to podocytes in DN is due to hyperglycemia or their loss is secondary to the glomerular damage [19,20] , in any case podocyte loss is a reliable marker for kidney damage. On the other side, there is no doubt that the accelerated formation (and later accumulation) of advanced glycation end products (AGEs) is associated with the progression of diabetes complications [21]. This relationship is usually referred to as a vicious circle (but rather a downward spiral towards increasingly serious diabetic complications) and such is the case of DN where increasing levels of AGEs play a causative role and at the same time, the impairment of renal function favors AGEs accumulation [22]. Although there are many formation pathways, endogenous AGEs are mainly formed by the Maillard reaction as part of physiological metabolism and normal aging, but this formation is increased in hyperglycemia. AGEs formation is due to the nucleophilic addition of the N-terminal end of a biomolecule (an amino acid generally, mainly lysine and arginine[23]) and a reducing sugar (mainly but not limited to, glucose), forming a Schiff base, after a more stable rearrangement, ketoamines known as Amadori products are formed. As the last step, through several reactions including dehydration, cyclization, fragmentation, and oxidation, the irreversible compounds known as AGEs are formed.[24] Although AGEs are a complex group of compounds there are specific AGEs that have a better correlation with some diabetic complications, among them, pentosidine, a fluorescent AGE formed with lysine and arginine, first described by Sell and Monnier [25], has been related to diabetic nephropathy[26], particularly free pentosidine in urine (uPen) has been determined as a marker of progression rate[27,28]. Besides, pentosidine has been associated with low estimated GFR in non proteinuric type 2 diabetic patients, issue that highlights pentosidine determination potential as CKD progression marker even in early stages[29]. Analytical control of urinary AGEs before and after pharmacological treatment provides valuable data for better understanding the association between hyperglycemia and diabetes complications. Indeed, the increased concentrations of uPen in diabetic and DN patients as compared to healthy individuals were reported in several studies [30] whereas after pharmacological interventions, the decrease of uPen was observed [22,27].
Another important issue is that hyperglycemia may induce angiotensin II expression in the podocytes by overregulating the angiotensinogen expression [31]. Angiotensin II production in the podocytes may also be activated by the proteinuria [32]. Angiotensin II leads to an increase of urinary protein excretion in the rat isolated perfused kidney [33]. Studies done in intact freshly isolated glomeruli suggest that angiotensin II depolarizes podocytes directly by opening a Cl- conductance, mediated by an AT1 receptor [34]. In culture, podocytes contain an intrinsic renin-angiotensin system, being able to form angiotensin II [35].
In vivo studies have shown that high-dose Angiotensin Receptor Blockers (ARBs) may induce regression of sclerosis, and regenerate the capillary loops [31]. A strict angiotensin blockade prevents the increase of intrarenal angiotensin II and podocyte abnormalities in type 2 diabetic rats with microalbuminuria [32,33].
The renin-angiotensin system (RAS) inhibition, reduces the leakage of proteins from the glomerular filtration barrier, but a RAS blockade increment in proteinuric patients usually entails a high rate of severe side effects[36], converting the search for novel therapeutic targets to achieve RAS inhibition without deleterious side effects in a main research field in the years to come [37]. Angiotensin converting enzyme inhibitors (ACEI) as well as ARBs decrease proteinuria and slow the progression of DN by mechanisms that cannot be attributed solely to effective control of systemic hypertension [34,35,38]. Nakamura et al, showed that after two months of Trandolapril treatment, albuminuria as well as the number of urinary podocytes decreased significantly in DM2 patients[35]. Barnett et al compared the effect of Telmisartan versus Enalapril in DM2 patients with nephropathy during five years showing that the effect in preventing the decline in the glomerular filtration rate (GFR) was similar for the two drugs [39].
While there are studies showing the effectiveness of ACEIs in podocyte injury [35] we have not found studies on the effect of ARBs over uPen levels and podocyte excretion. Furthermore, studying the effect of Telmisartan on podocyturia, AGEs levels, proteinuria, and metabolic control on early DM2 patients can contribute in clearing some of the mechanisms on the progression of the DN, and its treatment.
Subjects and Methods
The study was approved by the Ethics Committee of the University of Guanajuato, in adherence with the Declaration of Helsinki. Patients signed a letter of informed consent.
All patients continued with the treatment for diabetes prescribed at their clinical attention unit. Thirty patients, 5 males, 25 females with type 2 diabetes mellitus were recruited in the facilities at the Medical Research Department of University of Guanajuato, with an age range from 40 to 62 years old, with normal blood pressure, without systemic or urinary infections, or other chronic degenerative diseases. Subjects were not currently or previously treated with ACEIs or ARBs.
Clinical history and anthropometric parameters were recorded. After 10 hours fasting blood samples were taken. Serum glucose was determined by the glucose oxidase method (Spinreact, Spain), concentration of HbA1C was measured by chromatographic-spectrophotometric ion exchange analysis (Biosystems, Spain). Cholesterol, triglycerides and HDL levels were measured by an enzymatic spectrophotometric method (Spinreact SA Barcelona Spain). Kidney disease was evaluated based on K/DOQI criteria using serum creatinine. GFR was calculated by the formula of the MDRD study [4]. Albuminuria was assessed using Hemo-Cue (HemoCue AB, Angelholm Sweden). First morning urine for 5 consecutive days was collected.
Experimental
Urinary podocyte excretion was evaluated as reported by Hara et al [40,41] with modifications. For each patient, 5 ml aliquots of the urine collected in five days were pooled; 10 mL were then centrifuged (700 g, 5 min at 4°C, Eppendorf centrifuge 5810-R) and the obtained sediment was washed twice with phosphate buffered saline (PBS, 15 mM phosphate, 154 mM NaCl, pH 7.2) repeating centrifugation. Cellular pellet was fixed with 4% paraformaldehyde-0.5% glutaraldehyde. The cells were permeabilized with Triton X-100 at 0.1% for 3 min at 4 °C and washed once with PBS (700 g, 5 min at 4°C). Cellular pellet was incubated for 30 min at 4 °C in PBS containing fetal 10% bovine serum (FBS, heat inactivated). Subsequently, pellet was washed once with PBS and an aliquot incubated 1 h with the first antibody anti-human podocalyxin 1:10 (PHM-5) (Australian Monoclonal Development, Australia). Another aliquot (negative control) was incubated in the same conditions only with PBS. After washing with PBS, both cellular pellets were incubated for 1 h at room temperature with secondary antibody 1:100 (anti-mouse IgG-FITC, Cappel/ICN Biomedicals) with 10% FBS. Both samples were mounted on a slide with Vector mounting medium containing DAPI (Vector Mounting Medium with DAPI for fluorescence, VECTASHIELD) which stains the nucleus. Podocytes were observed using an epifluorescence microscope (Nikon HFX-II) to FITC using the filter B (Exc 450-490 nm) and DAPI using the UV filter (Exc. wavelength 320-380 nm).
Subjects with podocyturia received Telmisartan 80 mg once a day for two months. Patients were assessed clinically every two weeks. Adherence to treatment was verified by counting remaining tablets in the package. After treatment all laboratory assays were carried out again and a second quantification of urinary podocytes was done.
The determination of pentosidine (UPen) before and after Telmisartan treatment was carried out by ion-pair reversed phase liquid chromatography with fluorimetric detection (HPLC-FLD). Each urine sample (200 μL) was centrifuged at 1000 × g for 10 min, the obtained supernatant (160 μL) was treated with 40 μL heptafluorobutyric acid (HFBA 100 mM) and the precipitate was eliminated by centrifugation. Prior to injection into HPLC system (25 μl), the samples were appropriately diluted (when necessary to fit calibration curve). An Agilent series 1200 liquid chromatographic system equipped with a fluorimetric detector and a ChemStation (Agilent Technologies, Palo Alto, CA, USA) was used; the chromatographic column was a Zorbax Eclipse AAA (150 x 3.0 mm, 3.5 μm) from Agilent Technologies. Baseline separation of pentosidine (retention time 13.98 ± 0.08 min) was achieved using gradient elution with 1-20% acetonitrile and 3-30% methanol while keeping HFBA concentration in the mobile phase constant (20 mM); the column flow rate was 0.6 mL/min and the total chromatographic run 15 min. Fluorimetric detection was set at 325 nm and 385 nm for excitation and emission, respectively. Synthetic pentosidine standard was prepared and used throughout. The synthesis was carried out as reported elsewhere [42] with slight modifications. In brief, 150.1 mg of ribose (purity 99%), 246.3 mg of N-tBOC-lysine (purity 99%) and 274.3 mg of N-tBOC-Arginine (purity 95%) were dissolved in 2.5 mL of 0.5 M phosphate buffer (pH 8.0), and incubated for six days at 65° C. The two amino acids were protected with a t-butyloxycarbonyl (t-BOC) group on α-nitrogen to avoid participation of this amino group in the reaction; t-BOC groups were removed from the product by acid hydrolysis (trifluoroacetic acid 2M), the mixture was evaporated to dryness (SpeedVac, 60-70° C, 2h) and re-dissolved in 1.5 mL deionized water. Pentosidine purification was carried out by preparative chromatography using TSK-gel GMPVXL Tosohaas column (300 x 7.8 mm, 13 μm), formic acid 10 mM as a mobile phase and total flow rate 0.7 mL/min. Pentosidine column fraction (12.2 - 15.0 min) was collected, evaporated and re-dissolved in1.5 mL deionized water. The identity of pentosidine was confirmed by UV/Vis spectrophotometry, according to Slowik-Zylka et al.[43] Based on molar absorptivity coefficient assessed in the cited work (λmax = 315 nm, ε = 4522 L / (mol • cm), molar concentration of the obtained pentosidine standard was 87 mM. The stock solution was appropriately diluted to cover the calibration range 0.1-20 nM calibration range, the procedure was suitable for the determination of pentosidine at sub-nM concentrations (on-column detection limit 0.02 nM).
Statistical Analysis
Mean and standard deviation (mean ± SD), were obtained. Variables that did not fit to normal distribution are reported as median (range). Student’s T test or Mann-Whitney U test were carried out before Telmisartan treatment to compare differences between patients with and without podocyturia. After Telmisartan administration a T-test for dependent samples was done to compare results before and after treatment or a Wilcoxon test if the distribution was not normal. Significance was accepted for p values < 0.05. Statistics 6.0 (StatSoft, USA) program was used.
Results
Table 1 shows the clinical characteristics of the 30 patients. Blood pressure was normal. Serum glucose, HbA1c, cholesterol, and triglycerides values were similar in the patients with and without podocyturia. Only two patients with podocyturia had microalbuminuria over 20 mg/ml. We counted only non-overlapping podocytes with an intact nucleus, and with a bright fluorescent halo (podocalyxin positivity). A sample of podocytes is shown in fig. 1. Table 2 shows the clinical and biochemical data of patients before and after the Telmisartan treatment. After treatment there was a significant decrease in podocytes number (p<0.005). Serum cholesterol and LDL cholesterol levels decreased significantly after the Telmisartan treatment.
Table 1 General Characteristics of The Patients with And Without Podocyturia.
| Patients with podocyturia n= 10, M/F= 2/8 |
Patients without podocyturia n= 20, M/F= 3/17 |
p | |
|---|---|---|---|
| Age (years) | 50.5 ± 6.7 | 50.8 ± 6.5 | 0.9 |
| BMI (kg/m2) | 29.6 ± 4.4 | 28.4 ± 5.2 | 0.5 |
| SBP (mmHg) | 120.0 ± 8.2 | 117.0 ± 7.3 | 0.3 |
| DBP (mmHg) | 76.4 ± 4.8 | 77.1 ± 5.3 | 0.7 |
| Years since diagnosis of DM | 11.9 ± 4.1 | 9.7 ± 5.2 | 0.2 |
| Serum Glucose (mmol/L) | 9.69 ± 4.37 | 8.82 ± 3.09 | 0.5 |
| HbA 1C (%) | 9.3 ± 2.1 | 8.8 ± 1.6 | 0.5 |
| Serum Cholesterol (mmol/L) | 5.93 ± 1.32 | 5.66 ± 1.18 | 0.5 |
| Serum Triglycerides (mmol/L) | 2.06 ± 1.07 | 2.17 ± 1.28 | 0.8 |
| Serum Creatinine (mmol/L) | 77.79 ± 8.84 | 73.37 ± 7.07 | 0.1 |
| eGFR (ml/s) | 1.27 ± 0.14 | 1.36 ± 0.18 | 0.1 |
| Microalbuminuria (mg/l) | 6 (6- 114) | 5 (5- 18) | 0.4 |
| Podocyte Number | 6.5 ± 5.8 | 0.0 | 0.0000 |
M = males, F= females, Mean ± SD, Median (range), BMI= Body mass index, SBP= systolic blood pressure, DBP= diastolic blood pressure, HbA1c= Glycated hemoglobin, MAlb= Microalbuminuria (mg/l), eGFR= Estimated glomerular filtration rate.
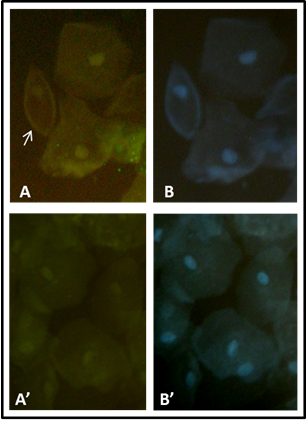
Fig.1. Urinary Podocytes stain. A, Urine sample with podocytes, positivity to podocalixin is showed by fluorescent halo (arrow). B, nuclei stain with DAPI. A’, Urine sample without podocytes. B’, nuclei stain with DAPI.
Table 2 Clinical and biochemical characteristics of the patients before and after Temisartan administration.
| Before n= 10 |
After n= 10 |
p | |
|---|---|---|---|
| SBP (mmHg) | 120 ± 8.2 | 119 ± 9.9 | 0.7 |
| DBP (mmHg) | 76.4 ± 4.8 | 76 ± 5.2 | 0.8 |
| Serum Glucose (mmol/L) | 9.69 ± 4.37 | 9.62 ± 4.29 | 0.9 |
| HbA 1C (%) | 9.28 ± 2.0 | 8.8 ± 1.3 | 0.4 |
| Serum Cholesterol (mmol/L) | 5.93 ± 1.32 | 5.00 ± 1.08 | 0.008 |
| Serum Triglycerides (mmol/L) | 2.06 ± 1.07 | 1.82 ± 0.85 | 0.3 |
| LDL Cholesterol (mmol/L) | 4.00 ± 0.92 | 2.98 ± 0.66 | 0.001 |
| Microalbuminuria (mg/l) | 6 (5 - 114) | 5.5 (5 -150) | 0.7 |
| Serum Creatinine (mmol/L) | 77.79 ± 8.84 | 81.3 ± 11.49 | 0.2 |
| eGFR (ml/s) | 1.27 ± 0.14 | 1.21 ± 0.21 | 0.2 |
| Podocyte Number | 4 (1 - 17) | 0 (0 - 8) | 0.005 |
| Podocytes/ml of urine | 0.045 (0.01-0.026) | 0.000 (0.000-0.06) | 0.005 |
| *Pentosidine (nM) | 2.66 ± 2.31 | 1.52 ± 0.85 | 0.0125* |
Mean ± SD, Median (range). SBP=Systolic blood pressure, DBP=Diastolic blood pressure, HbA1c= Glycated hemoglobin, eGFR = Estimated GFR,
*n=8.
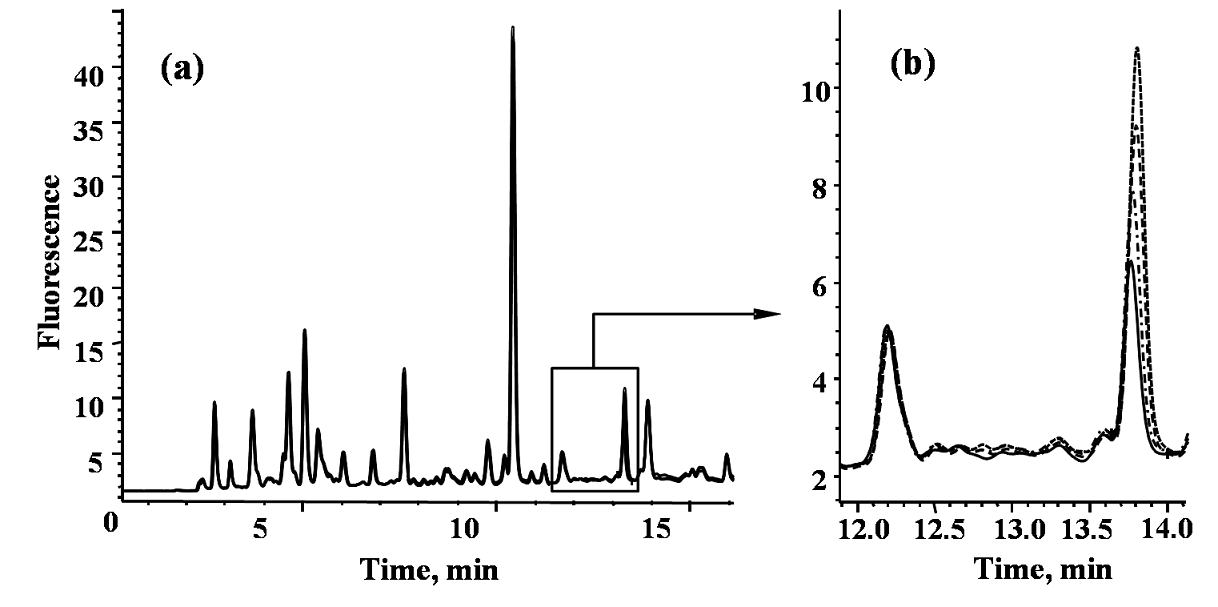
The chromatographic procedure reported previously [44] had been adopted for uPen determination; slight modification of the elution conditions was needed in order to assure baseline separation of pentosidine from other fluorescent sample components. In Fig. 2, typical HPLC-FLD chromatograms obtained analyzing the sample from healthy volunteer are presented. The purity of pentosidine elution peak in time region 13.85-14.14 min (corresponding to the peak width) was 0.999, which demonstrates efficient analyte resolution. For accuracy checking, quantification of pentosidine in this same sample was performed by external calibration and by the method of standard addition. Three-point standard addition was applied with 0.1 nM, 0.15 nM and 0.25 nM pentosidine added (as referred to the solution injected on column, Fig. 2) and each solution was analyzed in triplicate. The analytical parameters for the proposed chromatographic procedure were the following: the resulting calibration equation (with r2= 0.99986): A= 51.15 c (nM) + 0.27, standard error for the slope: 0.02; standard error for the intercept: 0.34, and 0.03 nM as the detection limit. The concentration of pentosidine was 0.223 ± 0.024 nM as determined by external calibration and 0.212 ± 0.017 nM by standard addition method. Unpaired t-test showed no significant difference between these two results (p > 0.05), confirming acceptable accuracy. Using this procedure, pentosidine was determined in urine samples from eight subjects before and after telmisartan administration. Prior to pharmacological treatment, mean pentosidine concentration was 2.66 ± 2.31 nM (range 1.09 - 7.03 nM) whereas in post treatment samples mean value was 1.52 ± 0.85 nM (range 0.77 - 3.20 nM). The post treatment decrease of uPen was statistically significant (paired t-test, p = 0.01255). Noteworthy, in all urine samples from DN patients, uPen concentrations were higher as compared to those found in healthy volunteers.
Discussion
Urinary podocyte excretion seems to appear earlier and more frequently than markers of kidney damage, like eGFR and microalbuminuria in normotensive DM2 patients (table 2). We also present the results indicating that Telmisartan treatment effectively decrease podocyte loss via urinary excretion; this effect was accompanied by statistically significant post treatment decrease of uPen in DM2 patients.
Podocyturia appears at an early phase of glomerular damage [16,17,45,46] playing an important role in the progression of glomerular diseases [45] Podocytes can be identified by an external protein known as podocalyxin, prominently expressed in glomerular epithelial cells [47,48].
While Hara et al utilized a slide centrifuge to obtain the urine sediment from a one day urinary specimen [40,41] in this study an immunocytochemistry technique was standardized, using a conventional centrifuge to obtain the urinary sediment collected during 5 days and then fix it, allowing to perform a more accurate counting of the excreted podocytes. The presence of a bright halo on the cell membrane indicating podocalyxin positivity as reported by Kerjaschki et al. and Hara et al [41] was possible using epifluorescence microscopy. According to Nakamura et al, diabetic patients with microalbuminuria excrete 0.67 podocytes/ml of urine, and the administration of Trandolapril decreased the counting to 0.08 podocytes/ml [35]. In this work an excretion of 0.045 (0.01-0.026) podocytes/ml was found, that diminished to 0.000 (0.00-0.06) (p< 0.005) podocytes/ml after the administration of 2 months of 80 mg/day of Telmisartan. Although the difference on the basal number of podocytes between our study and that of Nakamura does not have a clear explanation, patients in this study had an apparent evolution longer than ten years, while in Nakamura’s study there are no data on the diabetes evolution time in those patients.
It is evident that podocyte loss occurs in type 2 diabetes, and is related to proteinuria, but the precise sequence of events cannot be determined and the cause-effect relationships are still poorly understood [11] Podocyturia and proteinuria can be separated experimentally, the onset of proteinuria parallels the onset of podocyturia in some nephritis models, but proteinuria persists in late disease stages, suggesting that the detection of proteinuria cannot distinguish between a late stage of disease and an ongoing injury, whereas podocyturia may be more specific for the active injury [16] Microalbuminuria is a known risk factor in the development of diabetic nephropathy [49,50] although its presence is not necessarily accompanied by podocyturia. In this study, only 6.6% of the 30 patients presented microalbuminuria, this is in concordance with Adler et al [49] who, in 5097 DM2 patients, found only 333 with microalbuminuria (6.5%).
ARBs protect against the nephropathy progression independently of the antihypertensive mechanism [51-54] Several investigators have shown the antiproteinuric effect of ARBs in either hypertensive or normotensive DM2 patients, with different degrees of chronic kidney disease, in this case no changes were found in the microalbuminuria of the patients. A possible explanation may be the small number of patients with microalbuminuria in the study, besides the short time of Telmisartan administration.
Hyperglycemia has been considered as the principal factor in the development of the renal structural and functional alterations in diabetic nephropathy [55]. Telmisartan acts as a partial agonist of PPAR-γ, which by influencing the gene expression involved in glucose metabolism may improve insulin resistance, reducing glucose, insulin and triglyceride levels in rats as well as in diabetic patients[56-59]. Probably because of the short time of Telmisartan administration serum glucose as well as HbA1c levels remain unchanged, however, despite this, a significant decrease in the serum cholesterol and LDL-C levels after the treatment were found, in agreement with Derosa et al, who observed a significant reduction of serum cholesterol, LDL, and triglycerides levels after 12 months of 40 mg/day of Telmisartan [59]. In another study published in 2007, Derosa et al administered 40 mg/day of Telmisartan, plus 4 mg rosiglitazone/day to DM2 patients during 12 months, those patients showed a significant reduction in total cholesterol and LDL plasma levels compared with baseline [60] Same results on the plasma lipid profile have been described in hypertensive patients receiving Telmisartan in different doses [61,62]. Patients in this study had never received treatment with ACEIs or with ARBs, and all of them were normotensive. As after the Telmisartan administration for 8 weeks blood pressure levels remain unchanged, there is no evidence to support a possible action of blood pressure on the UPE. Considering that the metabolic and blood pressure conditions did not change after treatment, the beneficial changes on podocyturia should be attributed to Telmisartan treatment.
The effects of ACEIs on the urinary podocytes number have been described in animal models as well as in DM2 patients with early and late nephropathy [63] showing that the UPE is confined to the active phase of the glomerular injury [16,17] and that it develops in the earlier phases of the kidney disease, because at that time, the number of podocytes attached to the basement membrane is greater.
This study shows the beneficial effects obtained by a short period of Telmisartan administration on the UPE and on the lipid profile. The results obtained suggest that the podocyte protection by ARBs even in the face of deficient metabolic control could be of importance in the prevention and progression of diabetic nephropathy.
As an additional marker for DM2 patients during the experiment, uPen was determined in eight subjects before and after pharmacological intervention. Pentosidine is considered a biomarker with potential utility in diagnosis of diabetic nephropathy [64] and indeed, all DM2 subjects in this work presented higher uPen as compared to a non-diabetic volunteer, in agreement with many earlier studies [65,66]. As to the mode of action, it has been proposed that accumulating AGEs (pentosidine among many others) induce oxidative damage in podocytes, thus being involved in the development and progression of diabetic nephropathy [67]. Specifically, activation of the renin-angiotensin-aldosterone system (RAAS) in podocytes through AGE/RAGE signaling pathway has been reported and RAAS inhibitors were shown to protect from podocyte injury [68]. The finding of decreased concentrations of pentosidine and fewer podocytes in urine after pharmacological treatment with Telmisartan is particularly relevant since it supports that ARBs are capable of suppressing AGEs formation [22,69,70] and supports evidences that AGEs play a causative role in podocyte damage/loss [67].
Conclusions
Even though this study was performed on a small group of patients it should be stressed that analytically reliable procedures were employed producing clear and encouraging results. It is remarkable that the simultaneous determination of two different markers for CKD can, at the very least, assess in a more efficient way the renal function in DN than proteinuria (the most widely used diagnostic tool for CKD) alone, each marker independently are able to assess the therapeutic outcomes of Telmisartan in DN, which suggest that the simultaneous determination of both markers can give reliable information on CKD progression, a research issue that can be translated to the clinic in a close future and fulfill the gap in the search for simple, reliable and useful methods to successfully diagnose renal function and predict the therapeutic outcomes in early stages of CKD despite its origin. On the clinical approach it should be noted that the observed post treatment decrement in urinary podocytes and uPen suggests that Telmisartan as an angiotensin receptor blocker, either might tackle the formation of AGEs thus protecting podocytes from oxidative damage, or by a different mechanism exert an inhibitory effect on pentosidine excretion parallel to its protective role on podocyturia. Further studies are in progress in order to confirm the observed effects in a larger group of DM2 patients and including suitable control group.

