











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
There is a worldwide demand for design and synthesis of organic compounds with biological and pharmaceutical activity [1] using readily available starting materials by one-pot multicomponent reactions (MCRs) [2]. MCRs have many advantages in comparison with classical reactions, such as easier isolation and purification, lower energy consumption, using green solvents, easy operation, and more productivity with excellent chemo- and regioselectivities [3].
Barbituric acid and uracil derivatives have an important role in medicinal chemistry because for their biological activities [4]. There are many multicomponent condensations using barbituric acid and uracil as starting materials for the rapid synthesis of heterocyclic of pyrido-, pyrrolo-, and pyrimido-pyrimidines with various biological and pharmaceutical application [5]. Examples of their use are as antimicrobial [6], acaricidal [7], anti-inflammatory [8], anticancer [9], analgesic [10], sedative [11], anticonvulsants [12] and anesthetic agents [13].
Recently tetra-n-butylammonium bromide (TBAB) has emerged as one of the most widely used phase transfer catalysts. It combines the lipophilicity required for an efficient phase transfer catalyst with the hydrophobicity necessary for efficient catalyst recovery [14]. This and some other related ionic liquids have considerable interest as potential ecofriendly reagents due to their low vapor pressure. These liquids dissolve in many inorganic and organic materials, are nonvolatile and nonflammable, possess high thermal, high ionic conductivity, and chemical stability. It has been successfully used in the solid-liquid or liquid-liquid phase-transfer alkylation for C-C and C-X bond formation. It has been used as an inexpensive, mild, water-tolerant, and environmentally compatible an efficient homogeneous catalyst in different organic transformations [15]. TBAB is also a cheap, simple to use, readily available ionic liquid and has inherent properties like non-corrosive nature environmental compatibility, and ease of reusability [16].
In continuation of our interest in the synthesis of new heterocyclic compounds via one-pot, multicomponent reactions [17-23], herein we report a convenient and rapid method for the synthesis of pyrrolo[2,3-d ]pyrimidine derivatives by one-pot three-component reactions using arylglyoxals 1a-d, 6-amino-1,3-dimethyluracil (2) and barbituric acid derivatives 3a-c in the presence of a catalytic amount of TBAB (5 mol%) at 50 °C in ethanol.
Results and Discussion
In attempting to develop a simple, one-pot and short reaction pathway for the synthesis of various heterocyclic compounds, we reported earlier the synthesis of pyrazolo[3,4-d )]pyridines, pyrazolo[3,4-b]qunolin-5-ones and pyrazolo[4',3':5,6]pyrido[2,3-d]pyridin-5,7-diones, benzo[H]thieno[2,3-b]quinolone-9-yl(aryl)methanones, 6,7-dihydro-1H-indole-4(5H)-ones, acridine-1,8(2H,5H)-diones, 1,3-dimethyl-2,4-dioxo-1,2,3,4,5,8-hexahydropyrido[2,3-d]pyrimidine-6-carbonitriles, pyrrolo[3,2-d]pyrimidines [17-23].
The reaction of arylglyoxal 1a with 2, and barbituric acid derivatives 3a and 3c in ethanol under reflux conditions to form the desired polyfunctionalized pyrrolo[2,3-d]pyrimidine derivatives 4e and 4i has been previously reported [24]. Herein we report a simpler and more efficient procedure for this reaction.
We found that the reaction of arylglyoxals 1a-d with 2 and barbituric acid derivatives 3a-c in the presence of TBAB (5 mol%) in ethanol at 50 °C, instead of using reflux conditions, afforded polyfunctionalized pyrrolo[2,3-d]pyrimidine derivatives 4a-l in high yields with no sign of any dihydropyrido[2,3-d:6,5-d']dipyrimidine derivatives 5a-l formation (Scheme 1).
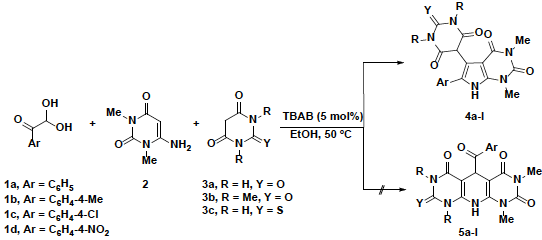
In our trial studies, the reaction of phenylglyoxal 1a, 2, and barbituric acid 3a was chosen as a model reaction (Table 1). First, we carried out this model reaction in the absence of a catalyst, but no product was observed even after 30 min of stirring at room temperature (Table 1, entry 1). To optimize the reaction temperature, the reactions were carried out at different temperatures ranging from room temperature to reflux. It was found that the yield was improved when the temperature was increased to reflux. Refluxing the reaction mixture in EtOH without any catalyst for 30 min gave the desired product 4a in 81% yield (Table 1, entry 3). Using 5 mol% of TBAB as organocatalyst for this reaction improved the yield (Table 1, entry 6), but using 10 mol% of TBAB for 65 min decreased the yield to 78% (Table 2, entry 9). The product was fully characterized using FT-IR (KBr), 1H-NMR and 13C-NMR spectral data, and by reference to the physical data, where appropriate [24].

Table 2 The effect of several catalysts in the synthesis of 4a.a
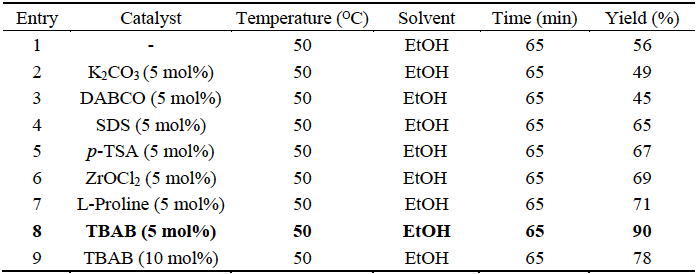
a Phenyglyoxal (1 mmol), barbituric acid (1 mmol) and 2 (1 mmol) in EtOH (5 mL).
To optimize the reaction temperature, the reactions were carried out at different temperatures ranging from room temperature to reflux. It was found that the yield was improved and the reaction time was shortened when the temperature was increased to 50 ºC.
The best result was obtained in terms of yield (90%) and reaction time (65 min) when the reaction was performed using 5 mol% of TBAB (Table 1, entry 6). To find the best solvent for this reaction, we repeated the model reaction in various solvents such as acetonitrile, dichloromethane (DCM), THF, DMF, MeOH, EtOH/H2O (1:1) and H2O. It was shown that EtOH was the best solvent in terms of yield and reaction time (Table 1, entry 6).
We also examined this reaction in the absence and presence of several other catalysts. When the reaction was carried out without any catalyst, the desired product 4a was formed in 56% yield (Table 2, entry 1). The use of bases like K2CO3 or 1,4-diazabicyclo[2.2.2]octane (DABCO) led to no improvement in yield, while acids, p-toluene sulfonic acid (p-TSA), ZrOCl2, sodium dodecyl sulfate (SDS), L-Proline gave small improvements (Table 2, entries 2-7). The best result was obtained when TBAB was used as a catalyst giving a yield of 90% (Table 2, entry 8).
To study the effect of the amount of catalyst, the reactions were carried out using different amounts of TBAB ranging from 5 to 10 mol%. The use of 5 mol% TBAB in EtOH led to optimum results. Using the larger amounts of TBAB did not improve the yields.
The reaction times (60-80 min), yields (73-95%) and melting points (>300 ºC) of synthesized pyrrolo[3,2-d]pyrimidine derivatives 4a-l using 5 mole% TBAB as catalyst are shown in Table 3. The substituted pyrrolo[3,2-d]pyrimidine derivatives 4a-l were characterized using FT-IR, 1H NMR and 13C NMR spectral data and microanalysis. In the 1H NMR spectra of products 4a-l, the characteristic singlets at around 11.79-12.29 and 4.55-5.32 ppm are attributed to the NH of pyrrole moiety and the CH of barbituric acid ring respectively. In the 13C NMR spectra of the products 4a-l, signals located around 151.1-174.2 ppm were ascribed to the three different carbonyl groups. In the FT-IR (KBr) spectra, the characteristic absorptions bands at 1671-1703, 1634-1689 and 1549-1589 cm-1 could be assigned to the vibrations of different carbonyl groups.
Table 3 Reaction conditions for the synthesis of pyrrolo[3,2-d]pyrimidine 4a-l. a
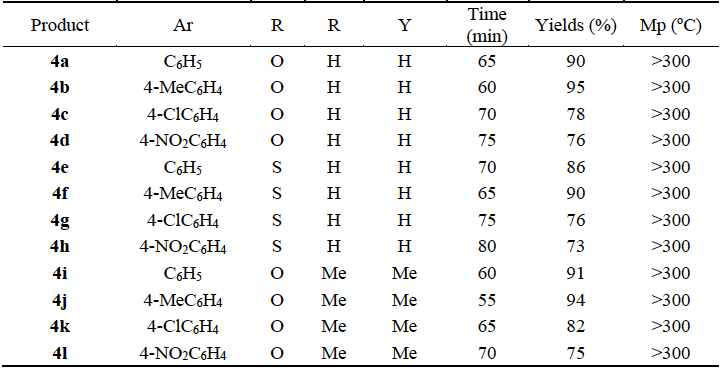
a Phenyglyoxal (1 mmol), barbituric acid (1 mmol) and 2 (1 mmol) in EtOH (5 mL), 50 ºC, 65 min.
A plausible mechanism for the formation of the products 4a-l is shown in Scheme 2. A sequence of reactions involving Knoevenagel condensation of 2 with aryglyoxals in the presence of TBAB forms the corresponding intermediate I. The observed products were then formed through an intermolecular condensation of intermediate I with barbituric acid derivatives with the ketone carbonyl group of I and loss of a water molecule, as shown in Scheme 2. It is expected that the use of TBAB in ethanol increases the stability of the ionized or tautomeric structures, thereby increasing the reaction rate and yields.

Conclusion
We have synthesized a new series of pyrrolo[2,3-d]pyrimidine derivatives 4a-l in high yields, by the one-pot three- component reaction of arylglyoxals 1a-d with 1,3-dimethyl-6-aminouracil 2 and barbituric acid derivatives 3a-c using 5 mol% TBAB as the catalyst in ethanol at 50 ºC. The simplicity of the method, ease of product isolation, mild reaction conditions, high yields, short reaction times and availability of the starting materials are the advantages of this procedure. Our method provides a simple synthesis of polyfunctionalized pyrrolo[2,3-d]pyrimidine derivatives with different substituents, which may have pharmaceutical and biological applications.
Experimental
The chemicals used in this work were purchased from Acros and Merck companies and were used without purification. Freshly distilled solvents were used throughout and anhydrous solvents were dried according to Perrin and Armarego [25]. Melting points were measured on an Electrothermal 9200 apparatus and are uncorrected. FT-IR (KBR) spectra were recorded on a Thermo Nicolet (Nexus 670) spectrometer using KBr discs. 1H (300 MHz) and 13C (75.5 MHz) NMR spectra were recorded on a Bruker DRX-300 Avance spectrometer in DMSO-d 6 with TMS as the internal reference. Elemental analyses were performed using a Leco Analyzer 932. The arylglyoxals were prepared as their hydrates by oxidation of the corresponding acetophenones with SeO2 [22].
General procedure for synthesis of pyrrolo[2,3-d]pyrimidine derivatives
A mixture of arylglyoxals 1a-d (1 mmol), 1,3-dimethyl-6-aminouracil (2) (155 mg, 1 mmol) and barbituric acid derivatives 3a-c (1 mmol), TBAB (16 mg, 0.05 mmol) in ethanol (5 ml) was stirred at 50 °C for the appropriate time as indicated in Table 3. The reaction was monitored by thin-layer chromatography TLC using (EtOAc/hexane, 5:1) as eluent. After completion of the reaction, the solid precipitate was filtered, washed with cold water and dried. Recrystallization from ethanol gave the desired products in 73-95% yields in Table 3.
5-(6-(Phenyl)-1,3-dimethyl-2,4-dioxo-2,3,4,7-tetrahydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)pyrimidine-2,4,6(1H,3H,5H)-trione (4a). White powder; 343 mg; mp >300 °C; FT-IR (KBr) νmax 3468, 3173, 3066, 2954, 1671, 1655, 1563, 1444, 1394, 1305, 1097, 1114, 1021, 979, 842, 748 cm−1; 1H NMR (DMSO-d 6 , 300 MHz) δ 11.85 (1H, s, exchanged by D2O addition, NH), 11.17 (2H, s, exchanged by D2O addition, 2×NH), 7.55 (2H, d, J = 6.9 Hz, Ar), 7.48 (2H, t, J = 7.2 Hz, Ar), 7.39 (1H, d, J = 6.6 Hz, Ar), 5.00 (1H, s, CH), 3.50 (3H, s, 3-Me), 3.15 (3H, s, 1-Me); 13C NMR (DMSO-d 6 , 75.5 MHz) δ 169.7 (C=O carbonyl), 159.1 (C=O carbonyl), 151.1 (C=O carbonyl), 139.7, 131.9, 130.9, 130.1, 129.7, 129.2 and 128.5 (aromatic C), 108.9 and 97.5 (pyrrole C), 48.1 (CH), 29.8 and 27.7 (CH3). Anal. Calcd for C18H15N5O5: C, 56.69; H, 3.96; N, 18.37. Found: C, 56.76; H, 4.11; N, 18.30.
5-(6-(4-Methylphenyl)-1,3-dimethyl-2,4-dioxo-2,3,4,7-tetrahydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)pyrimidine-2,4,6(1H,3H,5H)-trione (4b). White powder; 375 mg; mp >300 °C; FT-IR (KBr) νmax 3410, 3226, 3114, 2958, 1702, 1652, 1561, 1443, 1357, 1241, 1041, 974, 818 cm−1; 1H NMR (DMSO-d 6 , 300 MHz) δ 11.79 (1H, s, exchanged by D2O addition, NH), 11.16 (2H, s, exchanged by D2O addition, 2×NH), 7.42 (2H, bs, Ar), 7.29 (2H, bs, Ar), 4.96 (1H, s, CH), 3.47 (3H, s, 3-Me), 3.15 (3H, s, 1-Me), 2.34 (3H, s, Me); 13C NMR (DMSO-d 6 , 75.5 MHz) δ 169.7 (C=O carbonyl), 159.1 (C=O carbonyl), 151.0 (C=O carbonyl), 139.5, 131.9, 130.3, 129.8, 129.7, 129.0 and 128.4 (aromatic C), 108.5 and 97.4 (pyrrole C), 47.0 (CH), 31.1, 28.9 and 21.2 (CH3). Anal. Calcd for C19H17N5O5: C, 57.72; H, 4.33; N, 17.71. Found: C, 57.79; H, 4.25; N, 17.81.
5-(6-(4-Chlorophenyl)-1,3-dimethyl-2,4-dioxo-2,3,4,7-tetrahydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)pyrimidine-2,4,6(1H,3H,5H)-trione (4c). Pale yellow powder; 324 mg; mp >300 °C; FT-IR (KBr) νmax 3412, 3209, 2955, 1702, 1655, 1564, 1495, 1438, 1361, 1248, 1097, 1031, 977, 809, 505 cm−1; 1H NMR (DMSO-d 6 , 300 MHz) δ 11.91 (1H, s, exchanged by D2O addition, NH), 11.20 (2H, s, exchanged by D2O addition, 2×NH), 7.54 (4H, bs, Ar), 5.01 (1H, s, CH), 3.45 (3H, s, 3-Me), 3.14 (3H, s, 1-Me); 13C NMR (DMSO-d 6 , 75.5 MHz) δ 169.5 (C=O carbonyl), 159.1 (C=O carbonyl), 151.1 (C=O carbonyl), 139.6, 133.0, 130.6, 130.1, 129.7, 129.2 and 129.1 (aromatic C), 111.5 and 101.1 (pyrrole C), 46.9 (CH), 31.0 and 27.8 (CH3). Anal. Calcd for C18H14ClN5O5: C, 52.00; H, 3.39; N, 16.84. Found: C, 51.92; H, 3.45; N, 16.90.
5-(6-(4-Nitrophenyl)-1,3-dimethyl-2,4-dioxo-2,3,4,7-tetrahydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)pyrimidine-2,4,6(1H,3H,5H)-trione (4d). Orange powder: 324 mg; mp >300 °C; FT-IR (KBr) νmax 3442, 3392, 3139, 2963, 1642, 1589, 1438, 1328, 1438, 1328, 1218, 1148, 1118, 976, 580, 743, 706, 638 cm−1; 1H NMR (DMSO-d 6 , 300 MHz) δ 12.16 (1H, s, exchanged by D2O addition, NH), 11.28 (2H, s, exchanged by D2O addition, 2×NH), 8.26 (2H, d, J = 7.8 Hz, Ar), 7.74 (2H, d, J = 7.8 Hz, Ar), 5.17 (1H, s, CH), 3.44 (3H, s, 3-Me), 3.13 (3H, s, 1-Me); 13C NMR (DMSO-d 6 , 75.5 MHz) δ 169.3 (C=O carbonyl), 159.0 (C=O carbonyl), 151.2 (C=O carbonyl), 140.6, 132.7, 132.3, 129.7, 128.7, 128.3 and 127.4 (aromatic C), 111.9 and 98.2 (pyrrole C), 47.6 (CH), 31.1 and 27.8 (CH3). Anal. Calcd for C18H14N6O7: C, 50.71; H, 3.31; N, 19.71. Found: C, 50.63; H, 3.40; N, 19.81.
6-(Phenyl)-5-(4,6-dioxo-2-thioxohexahydropyrimidin-5-yl)-1,3-dimethyl-1,7-dihydro-2H-pyrrolo[2,3-d]pyrimidine-2,4(3H)-dione (4e). Pale pink powder: 341 mg; mp >300 °C (Lit.[24] >350°C); FT-IR (KBr) νmax 3354, 3178, 2257, 1648, 1550, 1439, 1300, 1219, 1154, 1006, 761, 696, 539 cm−1; 1H NMR (DMSO-d 6 , 300 MHz) δ 12.29 (1H, s, exchanged by D2O addition, NH), 11.94 (1H, s, exchanged by D2O addition, NH), 11.66 (1H, s, exchanged by D2O addition, NH), 7.41 (2H, d, J = 7.2 Hz, Ar), 7.26 (2H, t, J = 6.9 Hz, Ar), 7.16 (1H, d, J = 6.6 Hz, Ar), 5.20 (1H, s, CH), 3.44 (3H, s, 3-Me), 3.15 (3H, s, 1-Me); 13C NMR (DMSO-d 6 , 75.5 MHz) δ 173.8 (C=S), 161.3 (C=O carbonyl), 158.4 (C=O carbonyl), 151.9 (C=O carbonyl), 139.8, 132.1, 129.7, 129.5 and 129.2 (aromatic C), 106.8 and 100.2 (pyrrole C), 48.9 (CH), 30.9 and 27.8 (CH3). Anal. Calcd for C18H15N5O4S: C, 54.40; H, 3.80; N, 17.62. Found: C, 54.49; H, 3.71; N, 17.70.
6-(Methylphenyl)-5-(4,6-dioxo-2-thioxohexahydropyrimidin-5-yl)-1,3-dimethyl-1,7-dihydro-2H-pyrrolo[2,3-d]pyrimidine-2,4(3H)-dione (4f). Pale pink powder: 370 mg; mp >300 °C; FT-IR (KBr) νmax 3399, 3183, 3057, 2923, 2854, 1692, 1650, 1554, 1435, 1360, 1237, 1157, 1040, 1005, 875, 763, 743, 629, 534 cm−1; 1H NMR (DMSO-d 6 , 300 MHz) δ 12.28 (s, 1H, exchanged by D2O addition, NH), 11.75 (1H, s, exchanged by D2O addition, NH), 11.61 (1H, s, exchanged by D2O addition, NH), 7.21 (2H, bs, Ar), 6.96 (2H, bs, Ar), 4.66 (1H, s, CH), 3.36 (3H, s, 3-Me), 3.15 (3H, s, 1-Me), 2.31 (3H, s, Me); 13C NMR (DMSO-d 6 , 75.5 MHz) δ 173.6 (C=O), 161.6 (C=O carbonyl), 158.3 (C=O carbonyl), 151.0 (C=O carbonyl), 139.7, 132.2, 132.1, 129.6, 129.4 and 128.9 (aromatic C), 107.0 and 98.3 (pyrrole C), 49.0 (CH), 30.8, 29.7 and 23.5 (CH3). Anal. Calcd for C19H17N5O4S: C, 55.47; H, 4.16; N, 17.02. Found: C, 55.40; H, 4.26; N, 17.10.
6-(4-Chlorophenyl)-5-(4,6-dioxo-2-thioxohexahydropyrimidin-5-yl)-1,3-dimethyl-1,7-dihydro-2H-pyrrolo[2,3-d]pyrimidine-2,4(3H)-dione (4g). Pale pink powder: 328 mg; mp >300 °C; FT-IR (KBr) νmax 3359, 3195, 2959, 1648, 1549, 1433, 1378, 1296, 1221, 1152, 1006, 978, 787, 738, 668, 539 cm−1; 1H NMR (DMSO-d 6 , 300 MHz) δ 12.29 (1H, s, exchanged by D2O addition, NH), 12.06 (1H, s, exchanged by D2O addition, NH), 11.72 (1H, s, exchanged by D2O addition, NH), 7.43 (2H, d, J = 7.8 Hz, Ar), 7.39 (2H, d, J = 7.8 Hz, Ar), 4.68 (1H, s, CH), 3.45 (3H, s, 3-Me), 3.16 (3H, s, 1-Me); 13C NMR (DMSO-d 6 , 75.5 MHz) δ 174.0 (C=S), 161.9 (C=O carbonyl), 158.3 (C=O carbonyl), 152.0 (C=O carbonyl), 140.2, 131.7, 131.0, 128.7, 128.6 and 128.3 (aromatic C), 107.3 and 100.1 (pyrrole C), 49.0 (CH), 29.7 and 27.8 (CH3). Anal. Calcd for C18H14ClN5O4S: C, 50.06; H, 3.27; N, 16.22. Found: C, 50.13; H, 3.35; N, 16.13.
6-(4-Nitrophenyl)-5-(4,6-dioxo-2-thioxohexahydropyrimidin-5-yl)-1,3-dimethyl-1,7-dihydro-2H-pyrrolo[2,3-d]pyrimidine-2,4(3H)-dione (4h). Orange powder: 323 mg; mp >300 °C; FT-IR (KBr) νmax 3400, 3209, 2955, 1702, 1689, 1615, 1495, 1364, 1221, 1095, 827, 744, 514 cm−1; 1H NMR (DMSO-d 6 , 300 MHz) δ 12.21 (2H, s, exchanged by D2O addition, 2×NH), 11.86 (1H, s, exchanged by D2O addition, NH), 8.10 (2H, d, J = 7.8 Hz, Ar), 7.62 (2H, d, J = 7.8 Hz, Ar), 4.55 (1H, s, CH), 3.42 (3H, s, 3-Me), 3.14 (3H, s, 1-Me); 13C NMR (DMSO-d 6 , 75.5 MHz) δ 174.1 (C=S), 161.0 (C=O carbonyl), 158.2 (C=O carbonyl), 150.9 (C=O carbonyl), 141.1, 131.6, 127.5, 126.5, 125.0 and 122.8 (aromatic C), 110.4 and 100.9 (pyrrole C), 48.1 (CH), 31.0 and 27.8 (CH3). Anal. Calcd for C18H14N6O6S: C, 48.98; H, 3.08; N, 19.11. Found: C, 48.87; H, 3.19; N, 19.00.
5-(6-(Phenyl)-1,3-dimethyl-2,4-dioxo-2,3,4,7-tetrahydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (4i). White powder: 372 mg; mp >300 °C (Lit.[24] >350°C); FT-IR (KBr) νmax 3450, 3177, 2963, 1691, 1640, 1565, 1495, 1372, 1248, 1091, 1053, 975, 819, 742, 517 cm−1; 1H NMR (DMSO-d 6 , 300 MHz) δ 11.90 (1H, s, exchanged by D2O addition, NH), 7.35-7.65 (5H, m, Ar), 5.15 (1H, s, CH), 3.50 (3H, s, Me), 3.16 (6H, s, 2xMe), 3.13 (3H, s, Me); 13C NMR (DMSO-d 6 , 75.5 MHz) δ 168.1 (C=O carbonyl), 159.3 (C=O carbonyl), 152.4(C=O carbonyl), 151.1 (C=O carbonyl), 139.7, 131.6, 130.8, 129.6 and 128.5 (aromatic C), 109.2 and 97.3 (pyrrole C), 47.1 (CH), 31.1, 29.3 and 28.9 (CH3). Anal. Calcd for C20H19N5O5: C, 58.68; H, 4.68; N, 17.11. Found: C, 58.60; H, 4.77; N, 17.19.
5-(6-(4-Methylphenyl)-1,3-dimethyl-2,4-dioxo-2,3,4,7-tetrahydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (4j). White powder: 398 mg; mp >300 °C; FT-IR (KBr) νmax 3745, 3459, 2938, 1691, 1634, 1446, 1344, 1215, 1107, 1033, 905, 760, 631 cm−1; 1H NMR (DMSO-d 6 , 300 MHz) δ 11.85 (1H, s, exchanged by D2O addition, NH), 7.38 (2H, d, J = 7.5 Hz, Ar), 7.13 (2H, d, J = 7.5 Hz, Ar), 5.16 (1H, s, CH), 3.52 (3H, s, 3-Me), 3.33 (3H, s, 1-Me), 3.05 (6H, s, 1-Me, 3-Me), 2.26 (3H, s, Me); 13C NMR (DMSO-d 6 , 75.5 MHz) δ 167.8 (C=O carbonyl), 159.2 (C=O carbonyl), 152.3 (C=O carbonyl), 151.0 (C=O carbonyl), 140.7, 132.1, 129.7 129.3 and 129.3 (aromatic C), 112.1 and 100.6 (pyrrole C), 47.7 (CH), 31.2, 28.9, 27.9 and 21.2 (CH3). Anal. Calcd for C21H21N5O5: C, 59.57; H, 5.00; N, 16.54. Found: C, 59.45; H, 5.00; N, 16.62.
5-(6-(4-Chlorophenyl)-1,3-dimethyl-2,4-dioxo-2,3,4,7-tetrahydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (4k). Pale yellow powder: 364 mg; mp >300 °C; FT-IR (KBr) νmax 3392, 3220, 3108, 2951, 2875, 1703, 1651, 1562, 1443, 1359, 1241, 1038, 975, 816, 774, 702, 608, 502 cm−1; 1H NMR (DMSO-d 6 , 300 MHz) δ 11.95 (1H, s, exchanged by D2O addition, NH), 7.57 (4H, bs, Ar), 5.16 (1H, s, CH), 3.49 (3H, s, 3-Me), 3.16 (6H, s, 1-Me, 3-Me), 2.50 (3H, s, Me); 13C NMR: (DMSO-d 6 , 75.5 MHz) δ 168.0 (C=O carbonyl), 159.2 (C=O carbonyl), 152.4 (C=O carbonyl), 139.9, 133.1, 130.4, 130.1, 129.6 and 129.3 (aromatic C), 109.7 and 101.0 (pyrrole C), 47.2 (CH), 31.1, 30.4 and 28.9 (CH3). Anal. Calcd for C20H18ClN5O5: C, 54.12; H, 4.09; N, 15.78. Found: C, 54.20; H, 4.16; N, 15.70.
5-(6-(4-Nitrophenyl)-1,3-dimethyl-2,4-dioxo-2,3,4,7-tetrahydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (4l). Orange powder: 341 mg; mp >300 °C; FT-IR (KBr) νmax 3178, 3069, 2940, 1700, 1670, 1581, 1514, 1444, 1341, 1109, 979, 860, 752, 706 cm−1; 1H NMR (DMSO-d 6 , 300 MHz) δ 12.15 (1H, s, exchanged by D2O addition, NH), 8.34 (2H, d, J = 8.4 Hz, Ar), 7.80 (2H, d, J = 8.4 Hz, Ar), 5.32 (1H, s, CH), 3.51 (3H, s, 3-Me), 3.15 (6H, s, 1-Me, 3-Me), 2.50 (3H, s, Me); 13C NMR: (DMSO-d 6 , 75.5 MHz) δ 167.8 (C=O carbonyl), 159.2 (C=O carbonyl), 152.3 (C=O carbonyl), 151.0 (C=O carbonyl), 140.7, 132.0, 129.7, 129.6 and 129.3 (aromatic C), 112.1 and 102.7 (pyrrole C), 47.7 (CH), 31.2, 28.9 and 27.9 (CH3). Anal. Calcd for C20H18N6O7: C, 52.87; H, 3.99; N, 18.50. Found: C, 52.73; H, 4.05; N, 18.63.

