











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
Hydroxamic acids whether acyclic or cyclic, are interesting compounds which contain a hydroxyl group on a nitrogen atom of an amide or a lactam group. They are found to be associated with various activities such as anti-HIV 1, anti-Alzheimer diseases 2, in the treatment of metal poisoning 3, malaria 4, allergic diseases 5, and tuberculosis 6. Also, it has been reported that hydroxamic acids possess cytotoxic activity as result of the inhibition of the histone deacetylase (HDAC) enzyme 7. Some hydroxamic acids are presented in Fig.1, and these compounds exert significant cytotoxic activity in vitro, especially on prostate cancer cells 8),9),(10.

In the present work, we targeted the synthesis of several bicyclic hydroxamic acid derivatives, employing an uncommon nitro group rearrangement in the reaction between β-nitrostyrenes 11,12,13,14 and dimedone using a similar procedure to that reported by Larson et al 15. The cytotoxicity of the obtained compounds was evaluated to establish its potential anticancer activity.
Results and Discussion
Chemistry
The β-nitrostyrenes 2 used for the synthesis of hydroxamic acids 3, were prepared from a similar procedure 16, from nitromethane (1.1 equiv.), the corresponding benzaldehydes 1a-f (1.0 equiv.) and AcONH4 (0.4 equiv.) in glacial acetic acid under reflux for 2 h.
The intermediate nitrostyrenes (2a-f) were obtained in yields ranging from 91 to 95 % and their structures were determined by m.p., NMR and MS data. The β-nitrostyrenes 2a-f were reacted with dimedone in the presence of 2.2 equiv. of MeONa in dry MeOH, under nitrogen at 35-40 °C, yielding the target hydroxamic acids 3a-f (Scheme 1).
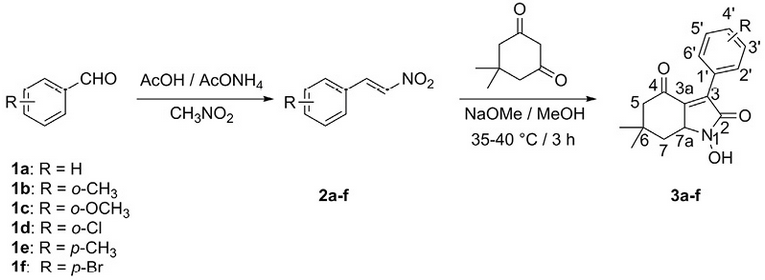
The mechanism whereby the hydroxamic acids 3a-f are formed is not clear; a proposal is shown (Scheme 2) supported by examples from literature 17), (18. The Michael addition of dimedone anion to nitrostyrene would lead to intermediate A, which in the presence of methanol and the methoxide ion would produce the nitrile oxide B, which undergoes a hydroxyl group attack toward the triple bond of the nitrile oxide producing intermediate C. Finally, the protonation and dehydration reactions of this intermediate gives the compound D which shows a tautomeric equilibrium with the hydroxamic acid 3a.
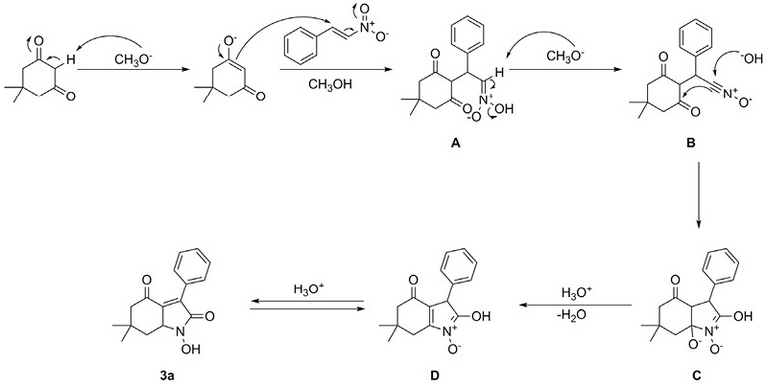
The chemical identification of products 3a-f was carried out using FTIR, 1H NMR, 13C NMR and mass spectral data. Finally, the hydrogenation reaction for compound 3a in the presence of Raney Nickel in ethanol produced compound 4, and subsequent dehydrogenation with Pd/C led to the pyrrole 5 (Scheme 3), confirming the structure of 1-hydroxy-6,6-dimethyl-3-phenyl-3,5,6,7-tetrahydro-1H-indole-2,4-dione (3a) and probably the heterocyclic framework of the other products, instead of 2-hydroxyiminofuran derivatives 19.Additionally, to corroborate the structure of pyrrole 5, it was prepared through a different synthetic route 20.
Cytotoxic activity
Cytotoxicity in vitro for compounds 3a-f was evaluated in six cancer cell lines, such as K562 (leukemia), HCT-15 (colon), MCF-7 (breast), PC-3 (prostate), U251 (CNS) and SKLU-1 (lung), using the Sulforhodamine B assay as is described by the National Cancer Institute of the United States of America 21. Preliminary screening with 50 µM solution of each compound allowed to determine that compounds 3a-f were more active in PC-3 and SKLU-1 cell lines. Then the concentration for tested compounds resulted 50 % inhibition of cell proliferation (IC50) was calculated. The results are summarized in the Table 1. In accordance with the IC50 values the compounds showed moderate cytotoxic activity against PC-3 and SKLU-1, among them 3e (IC50 = 23.38±0.9 μM against PC-3 and 9.31±0.1 μM against SKLU-1) compared to suberoylanilide hydroxamic acid (SAHA), a drug used in cancer chemotherapy, IC50 = 0.91±0.04 μM and 0.91±0.09 μM, respectively, another compound which showed good cytotoxic activity was 3f with IC50 values of 22.89±1.8 μM and 15.3±1.1 μM, for the respective cell lines (Tables 2, 3, 4, Figures 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12). This suggests that the presence of substituent on 4′ of the aromatic ring plays an important role in the cytotoxicity in comparison with the substituent located at 2′ as in 3b-d or when R is hydrogen (3a).
Table 1 Bicyclic hydroxamic acids and their cytotoxic activity in vitro against two cancer cell lines.
| Entry | R | Compound | Yield/% | IC 50 /μM | ||
|---|---|---|---|---|---|---|
| PC-3 | SKLU-1 | |||||
| 1 | H | 3a | 80 | 45.26±1.8 | 20.41±0.9 | |
| 2 | o-CH3 | 3b | 72 | >100 | 87.11±7.2 | |
| 3 | o-OCH3 | 3c | 75 | a | a | |
| 4 | o-Cl | 3d | 83 | 63.42±2.0 | 20.86±0.2 | |
| 5 | p-CH3 | 3e | 57 | 23.38±0.9 | 9.31±0.1 | |
| 6 | p-Br | 3f | 64 | 22.89±1.8 | 15.3±1.1 | |
| 7 | SAHA | 0.91±0.0.04 | 0.091±0.09 | |||
aNo activity
Table 2 Cytotoxic activity of SAHA.
| Cancer cell line | Concentration (µM) | Inhibition of the proliferation (%) | |||
|---|---|---|---|---|---|
| PC-3 | 1.00 | 87.76 | 86.85 | 95.40 | 91.80 |
| 3.10 | 73.98 | 75.37 | 74.80 | 72.20 | |
| 1.00 | 60.57 | 45.20 | 54.00 | 49.70 | |
| 0.31 | 29.51 | 35.37 | 23.30 | 32.70 | |
| SKLU-1 | 1.00 | 91.72 | 91.15 | 90.37 | 90.94 |
| 3.10 | 83.78 | 80.61 | 78.52 | 82.30 | |
| 1.00 | 59.36 | 59.28 | 52.84 | 56.01 | |
| 0.31 | 27.36 | 25.14 | 14.94 | 28.59 | |
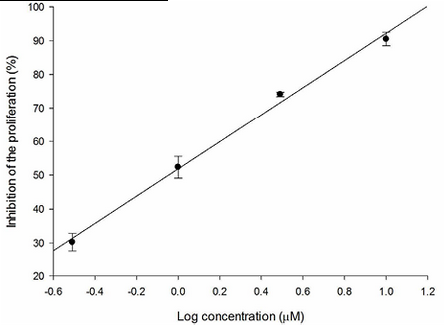

Fig. 3 Relation between SKLU-1 inhibition percentage and log of concentration for SAHA (R2 = 0.949).
Table 3 Cytotoxic activity of compounds 3a-f in PC-3.
| Compound | Concentration (µM) | Inhibition of the proliferation (%) | ||||
|---|---|---|---|---|---|---|
| 3a | 80.00 | 93.50 | 100.00 | 100.00 | 100.00 | * |
| 60.00 | 81.80 | 96.70 | 53.90 | 53.87 | * | |
| 40.00 | 23.30 | 41.70 | 30.06 | 34.57 | * | |
| 20.00 | 0.00 | 0.00 | 0.52 | 18.71 | * | |
| 3d | 100.00 | 100.00 | 100.00 | * | * | * |
| 80.00 | 63.60 | 76.30 | * | * | * | |
| 60.00 | 26.60 | 3.57 | * | * | * | |
| 40.00 | 13.20 | 15.70 | * | * | * | |
| 3e | 50.00 | 100.00 | 100.00 | 100.00 | 100.00 | * |
| 25.00 | 44.60 | 47.90 | 78.28 | 77.11 | * | |
| 12.50 | 0.00 | 9.58 | 0.00 | 4.52 | * | |
| 3f | 50.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 |
| 25.00 | 66.01 | 63.23 | 41.80 | 22.00 | 69.77 | |
| 12.50 | 17.30 | 20.46 | 4.08 | 0.00 | 10.87 | |
*Unregistered data
Table 4 Cytotoxic activity of compounds 3a-f in SKLU-1.
| Compound | Concentration (µM) | Inhibition of the proliferation (%) | ||
|---|---|---|---|---|
| 3a | 50.00 | 100.00 | 100.00 | 92.92 |
| 25.00 | 68.86 | 51.32 | 42.99 | |
| 12.50 | 10.07 | 22.69 | 26.95 | |
| 6.25 | 0.00 | 12.23 | 12.13 | |
| 3b | 100.00 | 82.09 | 82.51 | 94.04 |
| 75.00 | 39.10 | 20.28 | 67.15 | |
| 50.00 | 20.75 | 11.12 | 59.35 | |
| 25.00 | 2.47 | 0.00 | 16.88 | |
| 3d | 50.00 | 96.38 | 77.54 | 80.87 |
| 25.00 | 57.43 | 55.52 | 55.50 | |
| 12.50 | 19.67 | 28.92 | 24.38 | |
| 6.25 | 0.00 | 18.90 | 16.53 | |
| 3e | 25.00 | 98.59 | 95.07 | 95.90 |
| 12.50 | 46.88 | 48.36 | 50.43 | |
| 6.25 | 32.42 | 27.73 | 27.34 | |
| 3.12 | 20.74 | 19.19 | 18.30 | |
| 3f | 50.00 | 100.00 | 100.00 | 100.00 |
| 25.00 | 83.64 | 92.12 | 54.24 | |
| 12.50 | 31.60 | 41.64 | 35.56 | |
| 6.25 | 13.20 | 15.74 | 2.02 | |
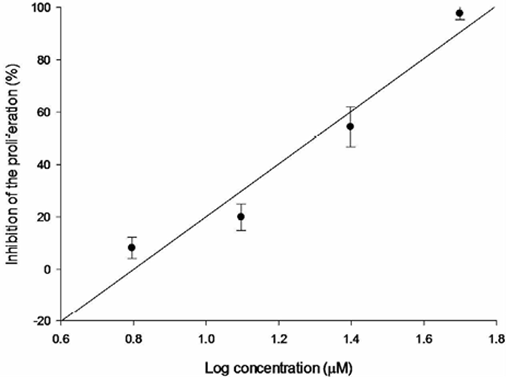
Fig. 4 Relation between SKLU-1 inhibition percentage and log of concentration for compound 3a (R2 = 0.946).

Fig. 5 Relation between PC-3 inhibition percentage and log of concentration for 3a compound (R2 = 0.952).

Fig. 6 Relation between SKLU-1 inhibition percentage and log of concentration for 3b compound (R2 = 0.859).

Fig. 7 Relation between PC-3 inhibition percentage and log of concentration for 3d compound (R2 = 0.828).
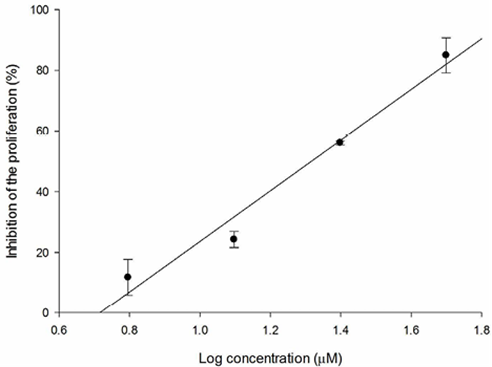
Fig. 8 Relation between SKLU-1 inhibition percentage and log of concentration for 3d compound (R2 = 0.972).

Fig. 9 Relation between PC-3 inhibition percentage and log of concentration for 3e compound (R2 = 0.985).
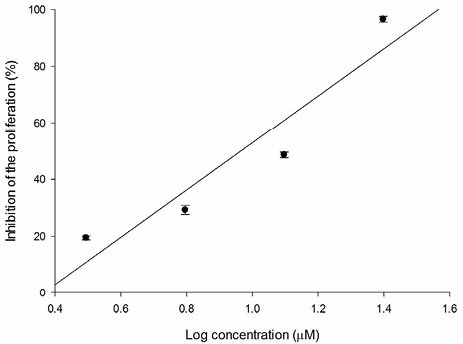
Fig. 10 Relation between SKLU-1 inhibition percentage and log of concentration for 3e compound (R2 = 0.891).
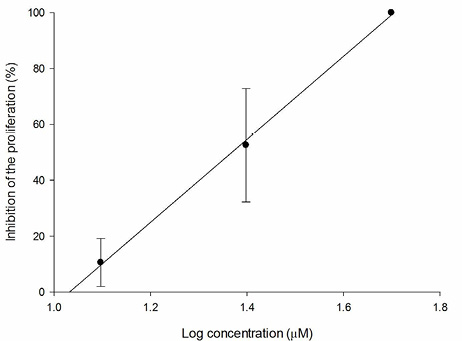
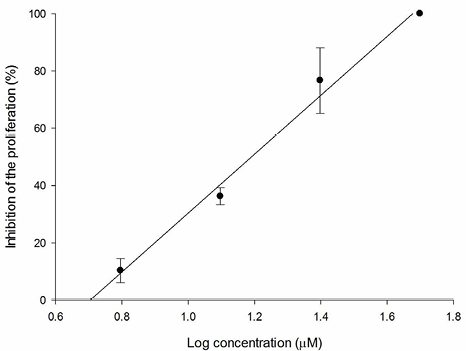
Fig. 11 Relation between PC-3 inhibition percentage and log of concentration for 3f compound (R2 = 0.998).
Conclusion
Six new bicyclic hydroxamic acids were synthesized in a two-step sequence starting from aromatic aldehydes 1. The β-nitrostyrenes 2 were obtained by Henry reaction of 1 with nitromethane followed by their reaction with dimedone in basic medium, through an uncommon reaction of nitro group, to obtain the products 3in good yields (57-80 %). The cytotoxicity of these compounds has been evaluated against different cancer cell lines. Thus, compounds 3e and 3f showed good cytotoxic activity.
Experimental
Materials
Starting materials and solvents were purchased from Sigma-Aldrich Co. and used without further purification. Melting points were determined using an Electrothermal Digital Melting Point Apparatus. IR spectra were measured using a Bruker Tensor 27 FT-IR spectrometer. 1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were recorded in a Jeol Eclipse 300 MHz spectrometer using CDCl3 as solvent and TMS as internal reference. Data for 1H NMR are recorded as follows: chemical shift (δ/ppm); multiplicity: s: singlet, d: doublet, dd: double doublet, t: triplet, td: triple doublet, m: multiplet or unresolved, bs: broad signal; the coupling constants are expressed in Hz. Data for 13C NMR are reported in terms of chemical shifts (δ/ppm). HR-EI-MS and HR-ESI-MS spectra were obtained in a Jeol M Station JMS-700 and a Jeol AccuTOF JMS-T-100 LC. Flash chromatography was performed with silica gel 60 (230-400 mesh ASTM). Silica gel 60 F254 aluminum plates were used for TLC analysis. Developing agents: phosphomolybdic acid 10 % in ethanol. The nitrostyrenes were synthesized following the reported procedure 16. All products were purified by recrystallization.
General procedure for the synthesis of compounds 2a-f.
Each benzaldehyde (4 mmol) was treated with nitromethane (4.4 mmol), ammonium acetate (1.6 mmol) in glacial acetic acid (5 mL). The mixture was stirred and refluxed for 2 h and the reaction mixture was cooled to 0 oC. The obtained products were filtered off and recrystallized from ethanol to give 2a-f. The 1H and 13C NMR spectra were in accordance to those cited in the literature for each described compound 12-14.
[(E)-1-(2-nitroethenyl)]benzene (2a).Isolated yield 92 %; yellowish solid; m.p.: 55-56 °C (from ethanol) (Ref. 12 55-57 oC, Ref. 13 59-62 oC). 1H NMR (300 MHz, CDCl3): δ= 7.43-7.32 (m, 2H, CHarom), 7.52-7.48 (m, 1H, CHarom), 7.57-7.53 (m, 2H, 2CHarom), 7.59 (d, J = 14 Hz, 1H, CH olefinic), 8.02 (d, 1H, J = 14 Hz, CH olefinic) ppm; 13C NMR (75 MHz, CDCl3): δ = 124.3, 129.6, 130.7, 131.5, 136.2, 139.5 ppm.
1-Methyl-2-[(E)-2-nitroethenyl]benzene (2b). Isolated yield 95 %; yellow solid; m.p.: 63-65 °C (from ethanol) (Ref. 13 112-118 oC, Ref. 14 yellow oil). 1H NMR (300 MHz, CDCl3): δ = 2.47 (s, 3H, CH 3 ), 7.27-7.24 (m, 2H, CH 2 arom), 7.39-7.35 (m, 1H, CHarom), 7.50-7.47 (m, 2H, CH olefinic, CH arom), 8.28 (d, J = 13.6 Hz, 1H, CH olefinic) ppm; 13C NMR (75 MHz, CDCl3): δ =20.1, 126.9, 127.5, 129.1, 131.6, 132.1, 136.9, 137.8, 139.4 ppm.
1-Methoxy-2-[(E)-2-nitroethenyl]benzene (2c). Isolated yield 94 %; yellow solid; m.p.: 60-62 °C (from ethanol) (Ref. 14 50-52 oC). 1H NMR (300 MHz, CDCl3): δ = 3.98 (s, 3H, OCH 3 ), 7.06-6.99 (m, 2H, 2CH arom), 7.50-7.46 (m, 2H, 2CH arom), 7.91 (d, J = 13.6 Hz, 1H, CH olefinic), 8.16 (d, J = 13.6 Hz, 1H, CH olefinic) ppm; 13C NMR (75 MHz, CDCl3): δ = 55.8, 111.6, 119.4, 121.3, 132.6, 133.6, 135.6, 138.5, 159.7 ppm.
1-Chloro-2-[(E)-2-nitroethenyl]benzene (2d). Isolated yield 91 %; brown solid; m.p.: 42-44 °C (from ethanol) (Ref. 12 40-42, Ref. 14 48-52 oC). 1H NMR (300 MHz, CDCl3): δ = 7.36-7.32 (m, 1H, CH arom), 7.43 (td, J = 7.7, J = 1.5 Hz, 1H, CH arom), 7.50 (dd, J = 8.0, J = 1.1 Hz, 1H, CH arom), 7.61-7.57 (m, 2H, CH olefinic, CH arom), 8.41 (d, 1H, J = 13.7 Hz, CH olefinic) ppm; 13C NMR (75 MHz, CDCl3): δ = 127.7, 128.7, 128.8, 130.9, 133.1, 135.3, 136.2, 139.1 ppm.
1-Methyl-4-[(E)-2-nitroethenyl]benzene (2e). Isolated yield 93 %; yellow solid; m.p.: 88-90 °C (from ethanol) (Ref. 12 100-102 oC, Ref. 13 102-106 oC, Ref. 14 106-108 oC). 1H NMR (300 MHz, CDCl3): δ = 2.44 (s, 3H, CH 3 ), 7.28 (d, J = 8.1 Hz, 2H, 2CH arom), 7.47 (d, J = 8.1 Hz, 2H, 2CH arom), 7.59 (d, J = 13.7 Hz, 1H, CH olefinic), 8.01 (d, J=13.7 Hz, 1H, CH olefinic) ppm; 13C NMR (75 MHz, CDCl3): δ = 21.9, 127.4, 129.4, 130.3, 136.5, 139.4, 143.3 ppm.
1-Bromo-4-[(E)-2-nitroethenyl]benzene (2f). Isolated yield 95 %; light yellow solid; m.p.: 115-118 °C (from ethanol) (Ref. 12 148-150 oC, Ref. (14) 153-155 oC). 1H NMR (300 MHz, CDCl3): δ = 7.40 (d, 2H, J = 8.4 Hz, 2CH arom), 7.60-7.55 (m, 3H, CH olefinic, 2CH arom), 7.93 (d, J = 13.7 Hz, 1H, CH olefinic) ppm; 13C NMR (75 MHz, CDCl3): δ = 127.0, 129.1, 130.6, 132.9, 137.6, 138.0 ppm.
General procedure for the synthesis of compounds 3a-f.
A solution of sodium methoxide (10.8 mmol) in anhydrous methanol (15 mL) was heated to 40 °C and 5,5-dimethylcyclohexane-1,3-dione (5.01 mmol) was added. To this solution were added the o-,p-substituted nitrostyrenes (2a-f) (5.01 mmol) and the mixture was stirred at 35-40 °C for 3 h, under nitrogen. Then, the mixture was cooled to 0 oC and acidified with 10 % hydrochloric solution until pH 3. The solid product was filtered off and recrystallized from hexane-acetone.
1-Hydroxy-6,6-dimethyl-3-phenyl-5,6,7,7a-tetrahydro-1H-indole-2,4-dione (3a). Isolated yield 80 %; beige crystals; m.p.: 145-148 °C (from hexane-acetone). 1H NMR (300 MHz, CDCl3): δ = 1.14 (s, 3H, CH 3 ), 1.17 (s, 3H, CH 3 ), 2.26 (d, J = 16.6 Hz, 1H, H-5α), 2.31 (d, J = 16.6 Hz, 1H, H-5β), 2.55 (dd, J = 2.8 Hz, J = 18.4 Hz, 1H, H-7α), 2.63 (dd, J = 1.4 Hz, J = 18.4 Hz, 1H, H-7β), 4.89 (dd, J = 2.1 Hz, J = 2.8 Hz, 1H, H-7a), 7.17-7.31 (m, 5H, 5H arom) ppm; 13C NMR (75 MHz, CDCl3): δ = 28.7, 28.8, 34.4, 36.9, 46.0, 51.3, 116.9, 127.6, 127.8, 128.9, 137.4, 158.6, 171.8, 193.2 ppm; IR: ῡ = 3251 (OH), 1699 (C=O), 1628 (C=O) cm-1; UV-Vis (methanol, c = 5.16.10-5mol dm-3): λmax (ε) = 207.0 (11793.72), 270.0 (10806.07) nm (mol-1 dm3 cm-1); MS (EI, 70 eV): m/z = 271 [M]+, 254, 187, 128, 115; HRMS (EI): m/z calculated for C16H17NO3 [M]+ 271.120843, found 271.1208.
1-Hydroxy-6,6-dimethyl-3-(2-methylphenyl)-5,6,7,7a-tetrahydro-1H-indole-2,4-dione (3b). Isolated yield 72 %; beige crystals; m.p.: 194-196 °C (from hexane-acetone). 1H NMR (300 MHz, CDCl3): δ = 1.15 (s, 3H, CH 3 ), 1.18 (s, 3H, CH 3 ), 2.25 (d, J = 16.2 Hz, 1H, H-5α), 2.29 (d, J = 16.2 Hz, 1H, H-5β), 2.45 (s, 3H, CH 3 arom), 2.57 (dd, J = 2.8 Hz, J = 18.3 Hz, 1H, H-7α), 2.65 (dd, J = 1.7 Hz, J = 18.3 Hz, 1H, H-5β), 5.07 (dd, J = 2.1 Hz, J = 2.8 Hz, 1H, H-7a), 6.9 (d, J = 7 Hz, 1H, CH arom), 7.08-7.15 (m, 3H, 3CH arom) ppm; 13C NMR (75 MHz, CDCl3): δ = 19.8, 28.7, 28.8, 34.4, 36.9, 42.8, 51.3, 117.4, 126.5, 127.5, 131.1, 136.2, 158.9, 171.6, 192.9 ppm; IR: ῡ = 3334 (OH), 1704 (C=O), 1634 (C=O) cm-1; UV-Vis (methanol, c = 4.35.10-5mol dm-3): λmax (ε) = 209.0 (16027.07), 270.5 (13451.70) nm (mol-1 dm3 cm-1); MS (EI, 70 eV): m/z = 285 [M]+, 268, 184, 183, 141, 115; HRMS (EI): m/z calculated for C17H19NO3 [M]+ 285.1365, found 285.1377.
1-Hydroxy-3-(2-methoxyphenyl)-6,6-dimethyl-5,6,7,7a-tetrahydro-1H-indole-2,4-dione (3c). Isolated yield 75 %; yellowish white crystals; m.p.; 192-193 °C (from hexane-acetone). 1H NMR (300 MHz, CDCl3): δ = 1.14 (s, 3H, CH 3 ), 1.15 (s, 3H, CH 3 ), 2.22 (d, J = 16.3 Hz, 1H, H-5α), 2.26 (d, J = 16.3 Hz, 1H, H-5β), 2.54 (dd, J = 2.8 Hz, J = 18.4 Hz, 1H, H-7α), 2.59 (dd, J = 2.1 Hz, J = 18.4 Hz, 1H, H-7β), 3.76 (s, 3H, OCH 3 ), 5.04 (dd, J = 1.7 Hz, J = 2.8 Hz, 1H, H-7a), 6.8-6.9 (m, 2H, 2CH arom), 7.1 (d, J = 6.0 Hz, 1H, CHarom), 7.2 (t, J = 7.0 Hz, 1H, CH arom) ppm; 13C NMR (75 MHz, CDCl3): δ = 28.3, 29.1, 34.4, 37.2, 42.2, 51.3, 55.8, 111.5, 116.1, 120.9, 125.5, 129.3, 129.7, 156.9, 159.0, 171.7, 193.0 ppm; IR: ῡ = 3317 (OH), 1710 (C=O), 1669 (C=O) cm-1; UV-Vis (methanol, c = 4.65.10-5mol dm-3): λmax (ε) = 204.5 (18863.64), 273.0 (13873.49) nm (mol-1 dm3 cm-1); MS (EI, 70 eV): m/z = 301 [M]+, 284, 228, 252, 115; HRMS (EI): m/z calculated for C17H19NO4 [M]+ 301.1314, found 301.1311.
3-(2-Chlorophenyl)-1-hydroxy-6,6-dimethyl-5,6,7,7a-tetrahydro-1H-indole-2,4-dione (3d). Isolated yield 83 %; beige crystals; m.p.: 142-144 °C (from hexane-acetone). 1H NMR (300 MHz, CDCl3): δ = 1.14 (s, 3H, CH 3 ), 1.17 (s, 3H, CH 3 ), 2.24 (d, J = 16.6 Hz, 1H, H-5α), 2.29 (d, J = 16.6 Hz, 1H, H-5β), 2.54 (dd, J = 2.8 Hz, J = 18.4 Hz, 1H, H-7α), 2.61 (dd, J = 1.7 Hz, J = 18.4 Hz, 1H, H-7β), 5.23 (bs, 1H, H-7a), 7.16-7.20 (m, 3H, 3CH arom), 7.32-7.35 (m, 1H, CH arom) ppm; 13C NMR (75 MHz, CDCl3): δ = 28.7, 28.9, 34.3, 36.9, 51.19, 115.6, 127.3, 129.6, 130.1, 133.4, 134.7, 157,5, 172.4, 193.0 ppm; IR: ῡ = 3321 (OH), 1707 (C=O), 1663 (C=O) cm-1; UV-Vis (methanol, c = 4.85.10-5mol dm-3): λmax (ε) = 205.0 (14718.53), 270.0 (11028.59) nm (mol-1 dm3 cm-1); MS (EI, 70 eV): m/z = 306 [M+H]+, 270, 252, 227, 196, 155, 115; HRMS (ESI): m/z calculated for C16H17 35ClNO3 [M+H]+ 306.08970, found 306.08917.
1-Hydroxy-6,6-dimethyl-3-(4-methylphenyl)-5,6,7,7a-tetrahydro-1H-indole-2,4-dione (3e). Isolated yield 57 %; orange-brown crystals; m.p.: 169-171 °C (from hexane-acetone); 1H NMR (300 MHz, CDCl3): δ = 1.14 (s, 3H, CH 3 ), 1.17 (s, 3H, CH 3 ), 2.26-2.29 (bs, 5H, CH 2 -5, CH 3 arom), 2.55 (dd, J = 2.5 Hz, J = 18.7 Hz, 1H, H-7α), 2.51-2.64 (m, 1H, H-7β), 4.85 (dd, J = 1.8, J = 2.5 Hz, 1H, H-7a), 7.08 (d, J = 7.8 Hz, 2H, 2CH arom), 7.11 (d, J = 7.8 Hz, 2H, 2CH arom) ppm, 13C NMR (75 MHz, CDCl3): δ = 21.2, 28.7, 28.8, 34.4, 36.9, 45.7, 51.3, 117.1, 127.4, 129.7, 134.4, 137.5, 158.9, 171.7, 193.1 ppm; IR: ῡ = 3235 (OH), 1706 (C=O), 1620 (C=O) cm-1; UV-Vis (methanol, c = 4.49.10-5mol dm-3): λmax (ε) = 206.0 (15014.40), 270.0 (12051.62) nm (mol-1 dm3 cm-1); MS (EI, 70 eV): m/z = 285 [M]+, 268, 255, 201, 115; HRMS (EI): m/z calculated for C17H19NO3 [M]+ 285.1365, found 285.1368.
3-(4-Bromophenyl)-1-hydroxy-6,6-dimethyl-5,6,7,7a-tetrahydro-1H-indole-2,4-dione (3f).
Isolated yield 64 %; yellowish-orange crystals; m.p.: 96-98 °C (from hexane-acetone). 1H NMR (300 MHz, CDCl3): δ = 1.09 (s, 3H, CH 3 ), 1.11 (s, 3H, CH 3 ), 2.18 (d, J = 16.6 Hz, 1H, H-5α), 2.24 (d, J = 16.6 Hz, 1H, H-5β), 2.51 (dd, J = 2.8 Hz, J = 18.4 Hz, 1H, H-7α), 2.54 (dd, J = 1.5 Hz, J = 18.4 Hz, 1H, H-7β), 4.82 (dd, J = 2.1, J = 2.8 Hz, 1H, H-7a), 7.09 (d, J = 8.5 Hz, 2H, 2CH arom), 7.43 (d, J = 8.5 Hz, 2H, 2CH arom) ppm, 13C NMR (75 MHz, CDCl3): δ = 28.5, 28.6, 34.4, 36.9, 44.9, 51.1, 116.6, 121.4, 129.4, 131.8, 134.5, 157.8, 172.2, 193.0 ppm; IR: ῡ = 3213 (OH), 1701 (C=O), 1645 (C=O) cm-1; UV-Vis (methanol, c = 3.78.10-5mol dm-3): λmax (ε) = 204.5 (14066.97), 269.0 (10100.72) nm (mol-1 dm3 cm-1); MS (EI, 70 eV): m/z = 349 [M]+, 334, 270, 253, 227, 155, 115, 83; HRMS (EI): m/z calculated for C16H16 79BrNO3 [M]+ 349.0314, found 349.0305.
Hydrogenation-dehydrogenation reactions of 3a to give products 4 and 5.
6,6-dimethyl-3-phenyl-1,2,5,6,7,7a-hexahydro-4H-indole-4-one (4). To a suspension of Raney Nickel (400 mg) previously activated and prehydrogenated, in dry ethanol (15 mL), the compound 3a was added (2.2 mmol) in dry ethanol (10 mL). The mixture was hydrogenated at 5 atm. until no more hydrogen is consumed. The reaction mixture was filtered on celite, the solution evaporated and the residue purified by silica gel column chromatography (hexane-ethyl acetate 90:10 as eluent), to give the product 4 (175 mg), 33 % yield; yellow crystals; m.p.: 232-234 °C (from hexane-ethyl acetate). 1H NMR (300 MHz, CDCl3): δ = 0.92 (s, 3H, CH 3 ), 1.04 (s, 3H, CH 3 ), 2.00 (s, 2H, CH 2 -7), 2.3 (s, 2H, CH 2 -5), 3.28 (dd, J = 4.8, J = 10.5 Hz, 1H, H-2α), 3.95 (t, J = 11.1 Hz, H-2β), 4.15 (dd, J = 4.8, J = 11.1 Hz, 1H, H-3), 7.19-7.32 (m, 5H, 5CH arom) ppm, 13C NMR (75 MHz, CDCl3): δ = 28.0, 28.8, 33.6, 36.8, 43.4,50.5, 55.3, 105.2, 125.8, 126.8, 127.6, 139.3, 146.1, 186.4 ppm; IR (KBr): ῡ = 3396 (NH), 1587 (C=O), 11481 (C=C) cm-1; MS (EI, 70 eV): m/z = 241 [M]+, 240, 184, 164, 157, 128, 83. HRMS (EI): m/z calculated for C16H19NO [M]+ 241.1466, found 241.1445.
6,6-dimethyl-3-phenyl-1,5,6,7-tetrahydro-4H-indole-4-one (5). The compound 4 (0.5 mmol) in toluene anhydrous (10 mL) was refluxed in the presence of 10 % Pd/C (10 mg) for 3 h. The reaction mixture was filtered on celite, the solution evaporated and the residue purified by silica gel column chromatography (hexane-ethyl acetate 60:40 as eluent), to give compound 5 (52 mg), 43 % yield;white crystals; m.p.: 245-247 °C (from hexane-ethyl acetate). 1H NMR (300 MHz, CDCl3): δ = 1.14 (s, 6H, 2xCH 3 ), 2.42 (s, 2H, CH 2 -5), 2.2.69 (s, 2H, CH 2 -7), 6.71 (s, 1H, H-2), 7.24 (t, J = 7.5 Hz, 1H, H-4´), 7.34 (t, J = 7.5 Hz, 2H, H-3´, H-5´), 7.64 (d, J = 7.5 Hz, 2H, H-2´, H-6´),7.35-7.63 (m, 5H, 5CH arom.) ppm, 13C NMR (75 MHz, CDCl3): δ = 28.6, 35.6, 37.4, 53.3, 115.9, 117.4, 125.0, 126.7, 128.0 128.7, 134.3, 144.0, 193.8 ppm; IR: ῡ = 3259 (NH), 1633 (C=O), cm-1; MS (EI, 70 eV): m/z = 239 [M]+, 183, 154, 125, 97, 84. HRMS (EI): m/z calculated for C16H17NO [M]+ 239.1310, found 239.1319.
Cytotoxicity evaluation
Cancer lines were obtained from the National Cancer Institute of the United States. Cytotoxicity data were determined using the protein-bound-sulforhodamine (SRB) assay in microculture; the assay measures cell proliferation as described by Monks method 21. Cell lines were cultivated in RPMI-1640 medium with 10 % (v/v) fetal bovine serum, 2 μM-glutamine, 100 IU/cm3penicillin G, 100 mg/cm3 streptomycin sulfate, and 0.25 μg/cm3 of amphotericin B (Gibco Grand Island, NY). The incubation was at 37 °C in a 5 %, CO2 atmosphere and 100 % relative humidity. After replication, cells were detached with a 0.05 % (w/v) trypsin-EDTA solution, counted in a hemocytometer, and diluted with complete medium to 5 x 103 cells/cm3 (K-562, MCF-7), 7.5 x 103 cells/cm3 (U-251, PC-3, and SKLU-1), or 10 x 103 cells/cm3 (HTC-15).
Microtiter plate wells were filled with 100 mm3 aliquots of cell suspension and incubated. After 24 h cells were treated with logarithmically diluted concentrations of sample compounds. Test samples were dissolved in DMSO to provide stock solutions (40 μM) and were then diluted to yield solutions of 100, 31, 10, 3.1 and 1 μM, for use in treatment of cell suspensions. After 48 h, cultures were fixed in situ by addition of 50 mm3 50 % (w/v) trichloroacetic acid and incubated for 60 min at 4 °C. Cells were harvested by centrifugation, washed three times, and dried. Cell pellets were suspended in 100 mm3 0.4 % (w/v) SRB solution in 1 % (v/v) acetic acid for 30 min. Unbound SRB was removed by washing with 1 % (v/v) acetic acid and protein-linked dye was extracted with 10 μM tris(hydroxymethyl)methaneamine solution, and quantitated by spectrometry at 515 nm. The IC50 values were calculated according to Monks 21.

