











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Step-growth polymers such as polyamides (PAs) with the imidazolium functionality are especially sparse in the literature. Baird and Dervan coupled imidazole or pyrrole groups with carboxylic acids to synthesize PAs through classical solid-phase peptide synthesis [1] and Sugiyama and Bando synthesized N-methylimidazole-containing polyimides (PIs) for sequence-specific DNA alkylation [2]. However, PAs and PIs are high-performance polymeric materials and application of these polymers is restricted as a result of not being processable due to limited solubility and high melting temperature. Therefore, researchers have focused on the development of structurally modified aromatic PAs and PIs with increased solubility and better processability through the incorporation of heterocyclic units, packing-disruptive flexible linkages and breaking symmetry and regularity of the polymer chain [3-10]. These approaches produce separation of chains, weakening of hydrogen bonding and lowering of chain packing with gaining free volume without affecting their thermal stability [11-16]. The rigidity based on the aromaticity and polarity of the heteroaromatic rings contributes to the thermal and chemical stability and retention of mechanical properties of the resulting polymer at elevated temperatures [17-19]. However, the polarizability resulting from the heteroatoms such as nitrogen atoms in the imidazole ring improves the polymer’s solubility in organic solvents [20-24]. Moreover, Imidazoles and more specifically 2,4,5-triarylimidazoles are known as inhibitors, herbicides and fungicides, therapeutic agents and plant growth regulators, [25-30] and their optical properties (chemiluminescence and fluorescence) are of particular concern for material scientists. The pollution of water resources due to the indiscriminate disposal of heavy metals has been causing worldwide concern for the last few decades. It is well known that some metals can have toxic or harmful effects on many forms of life [31-33]. Metals, which are significantly toxic to human beings and ecological environments, include chromium, lead, mercury, manganese, and cadmium, etc. The concentrations of some of the toxic metals are higher than permissible discharge levels in these wastewaters [34]. It, therefore, becomes necessary to remove these heavy metals from these effluents by an appropriate treatment before releasing them into the environment. Methods such as membrane filtration, ion exchange and electrolysis are not widely used because of their high cost and low feasibility. Among different technologies for removal of the heavy metal ions from aqueous solution, adsorption is a reliable and widely used method due to its simplicity and easy operational conditions [35,36]. Attention has been focused on the various adsorbents, which have metal-binding capacities and are able to remove unwanted heavy metals from contaminated water. Nowadays chelating polymers are increasingly used in the removal of metal ions due to their high adsorption capacities, selectivity and durability [37]. Chelating polymers are, in general, coordinating polymers containing one or more electron donor atoms (Lewis base) such as nitrogen, sulfur, oxygen, and phosphorus that can form coordinate bonds with most of the toxic heavy metals (Lewis acid) [38]. Imidazole heterocyclic ring and its derivatives such as lophine (substituted imidazole) are useful n-type building blocks with high electron affinity and good thermal stability and have been successfully incorporated in small molecules and polymers as an electron-transport component [39,40].
The main objective of this work was to synthesize a diamine with numbers of imidazole rings and its use in preparation of PAs and PIs having chains rich of imidazole rings.. These polymers are expected to show high thermal stability and photoluminescence properties due to presence of lophine structures in the backbone, and must also have good solubility in organic solvents as a result of presence of hetero-aromatic and ether linkages in the polymer chains. The target diamine was characterized by FT-IR, 1H and 13C NMR and used in step-growth polymerization with several commercial dicarboxylic acids and dianhydrides. The polymers were characterized by FT-IR and H NMR and their properties such as solubility, crystallinity, thermal stability, photophysical properties, and heavy metal ions removal from water were investigated.
Synthesis and Characterization of Monomer and Polymers
The objective of this study was the preparation of novel imidazole based polymers (PAs and PIs) by step-growth polymerization method from the target diamine IV. Scheme 1 illustrates synthesis steps of the target diamine (IV). Compounds (I) and (II) were synthesized by condensation of benzil with 2-chloro-5-nitrobenzaldehyde and ammonium acetate which is well known as a classical but convenient synthetic method for preparation of triaryl imidazole [19,41]. The dinitro (III) was synthesized by nucleophilic aromatic substitution reaction of compound II with compound I in DMAc and in the presence of K2CO3. The catalytic hydrogenation of dinitro III to the corresponding diamine IV was accomplished by means of hydrazine hydrate in ethanol in the presence of catalytic amount of palladium on activated carbon. The FT-IR spectrum, and 1H and 13C NMR spectra of diamine IV are shown in Figures 1, 2 and 3, respectively.




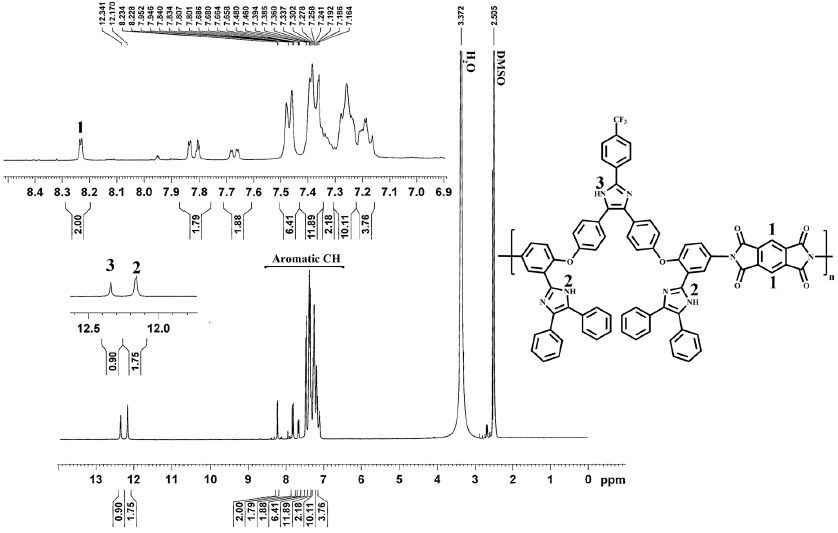
The nitro groups of compound III yielded two characteristic absorption bands at 1522 and 1350 cm−1 (-NO2 symmetric and asymmetric stretching, respectively). After reduction, these absorption peaks disappeared and the primary amino group in compound IV showed the typical absorption pair at 3484 and 3378 cm−1 as shown in Fig. 1. The signals at 4.47 ppm, and at 10.55 and 13.13 ppm are due to the protons of -NH2, and -NH of the imidazole rings, respectively. Aromatic protons in the region of 6.17-8.27 showed the expected multiplicity and integration values. These results and the assigned 13C NMR signals in the experimental section confirmed that the diamine IV prepared herein is consistent with the proposed structure. A series of novel PAs were prepared in good yields by direct polycondensation of diamine with three aliphatic and aromatic dicarboxylic acids in the presence of TPP and Py as condensing agent which is an efficient way to form amide bonds, as shown in scheme 2. All the PAs were characterized using 1H NMR, 13C NMR, FT-IR and elemental analysis techniques and the results were summarized in section 2.6.1. 1H NMR spectra of the representative PA1 and PA2 were illustrated in Fig. 4, where all the peaks could be readily assigned to the protons in the repeating unit. The proton of amide group was observed at 10.69-10.80 and 11.01-11.23 ppm for the aromatic PAs (PA2, PA3) and at 10.16-10.27 ppm for the aliphatic PA (PA1). Two novel PIs were prepared by polycondensation of equal molar amounts of the diamine monomer (V) with commercially available tetracarboxylic dianhydrides BCDA and PMDA, as shown in Scheme 2. The polycondensation was carried out in NMP solution at room temperature for 24 h to form poly(amic acid)s and followed by chemical imidization with acetic anhydride and pyridine at 130 ºC. The chemical imidization method has been chosen because the PIs obtained by this method usually exhibit better solubility than thermally cured ones. The poor solubility of thermally cured polyimide was attributed to partial crosslinking within polymer chains during thermal imidization [42]. The elemental analysis values generally agreed with the calculated values for the proposed structures of PIs. PIs were characterized using FT-IR, 1HNMR and elemental analysis techniques and the results were summarized in section 2.3.2. FT-IR spectrum of PI2 exhibited the characteristic absorption bands of the imide ring at 1779 cm-1 of asymmetric C=O stretching, 1712 cm-1 of symmetric C=O stretching and 1297 cm-1 (C-N stretching) as shown in Fig. 1. 1H NMR spectrum of the representative polyimide PI2 as an example is illustrated in Fig. 5.

Solubility
The solubility behavior of these new PAs and PIs was determined at concentration of 5% (W/V) in a number of solvents and the results are tabulated in Table 1. All PAs exhibited excellent solubility in polar aprotic solvents such as DMAc, NMP, dimethylsulfoxide (DMSO) and dimethylformamide (DMF) at room temperature. All PAs prepared in the mixture of TPP/NMP/Py/LiCl exhibited excellent solubility in polar aprotic solvents such as NMP, DMAc, DMF, DMSO and even in less polar solvents like pyridine, and m-cresol. The good solubility should be due to presence of different functional groups in the polymer backbone such as bulky heterocyclic pendant, ether linkage, -CF3 group, amide joints and unsymmetrical structure. These factors and also the amorphous nature contributed to the enhanced solubility of these PAs through better penetration of solvent molecules, formation of H-bonding and dipole-dipole interaction with polar molecules of organic solvents. In addition, the solubility varies depending upon the dicarboxylic acid used. The PAs which were synthesized from aliphatic dicarboxylic acids (PA1) exhibited better solubility behavior in less polar solvents such as Py and m-cresol. The presence of methylene units instead of rigid phenyl improves the solubility of these PAs.
Table 1 Solubility behavior of PAs and PIs
The solubility was determined by using 5% (w/v) concentration
++: Soluble at room temperature; + : Soluble on heating at 60 °C; − Insoluble on heating at 60 °C.
DMAc, N,N -dimethyl acetamide; DMF, N,N´-di-methyl formamide; NMP, N-methyl pyrrolidone; DMSO, dimethyl sulfoxide; THF, tetrahydro furan; Py, pyridin; CHCl3, Chloroform.
In general, PIs show lower solubility compared with PAs. These PIs were dissolved in high polar solvents, such as DMF, DMAc, DMSO, NMP at room temperature, and in moderate polar solvent such as Py and m-crosol by heating at 60 °C. The good solubility of these PIs might be attributed to the flexible ether and bulky groups such as phenyl, polar hetroaromatic imidazole rings and -CF3 pendants which prevent close chain-packing and allow the solvent molecules to diffuse into the polymer chains, and interact with polar groups in the polymer backbone. The solubility of these polymers is comparable, in spite of having many phenyl and bulky groups in the backbone, with the solubility of previously reported PIs [43]. In addition, the solubility varies depending upon the dianhydrides used. Polymers with flexible linkages are more soluble than those with rigid linkage. Therefore PI1 derived from more flexible dianhydride BCDA was so more soluble than aromatic PMDA in organic solvents.
X-Ray Diffraction of the PAs and PIs
The crystallinity of the prepared PAs and PIs as powder samples was examined by WAXD analysis with graphite-monochromatized Cu Kα radiation and with 2θ ranging from 0 to 50º. The WAXD patterns of these polymers showed an amorphous nature and the results were shown in Fig. 6. The amorphous nature of the PAs and PIs can be attributed to their unsymmetrical structural units and the bulky substituted imidazole and CF3 groups, which reduced the intra- and interpolymer chain interactions, resulting in loose polymer chain packaging and aggregates. Therefore, the amorphous nature of the resulting PAs and PIs would endow them a good solubility.

UV-Vis Absorption and Fluorescence Characteristics
The photophysical properties of the PAs and PIs were investigated by UV-visible and fluorescence spectroscopy in NMP solution. The absorption and fluorescence spectra of solution of the PAs and PIs (0.1 g/dL) are shown in Figures 7A and 7B, respectively, and the spectral data were listed in Table 2. These PAs and PIs showed strong UV absorption with the maximum wavelength at (λmax(ab)) 319-329 nm, which was assigned to π-π* transition resulting from the conjugation between the aromatic rings and nitrogen atom in the pendent group. The comparison of the absorption spectra of PAs and PIs show a discernible similarity demonstrating that the conjugated core is the absorbing unity. However, the absorption peaks of aromatic PA2, PA3 and PI2 is red-shifted and broadened compared to the absorption band of the PA1 and PI1, corresponding to the extended π-π* transitions of the polymer backbone. Fig. 7B shows the emission spectra of the PAs and PIs in dilute NMP solution. The excitation wavelength was 310 nm in all cases. The fluorescence emission spectra of the PAs exhibited peaks with maxima at 415-492 nm. Moreover, the emission intensity of aliphatic and aromatic PAs and PIs was not exactly the same, which confirms that the fluorescence intensity is affected by incorporation of alkyl and phenyl groups into the polymer main chain. To measure the photoluminescence (PL) quantum yields (Φf), dilute polymer solutions (0.2 g/dL) in NMP were prepared. A 0.10 N solution of quinine in H2SO4 (Φf= 0.53) was used as a reference [44]. The Φf values for PAs and PIs were in the range of 10-34%. The aliphatic polymers exhibited blue shift with higher quantum yield. The higher Φf of fluorescence emission of aliphatic PA1 and PI1 compared with the aromatic counterparts can be attributed to the effectively reduced conjugation and capability of charge-transfer complex formation by aliphatic diacids and dianhydrides with the electron-donating diamine moiety in comparison with that of the stronger electron-accepting aromatic diacids and dianhydrides.
Thermal Properties
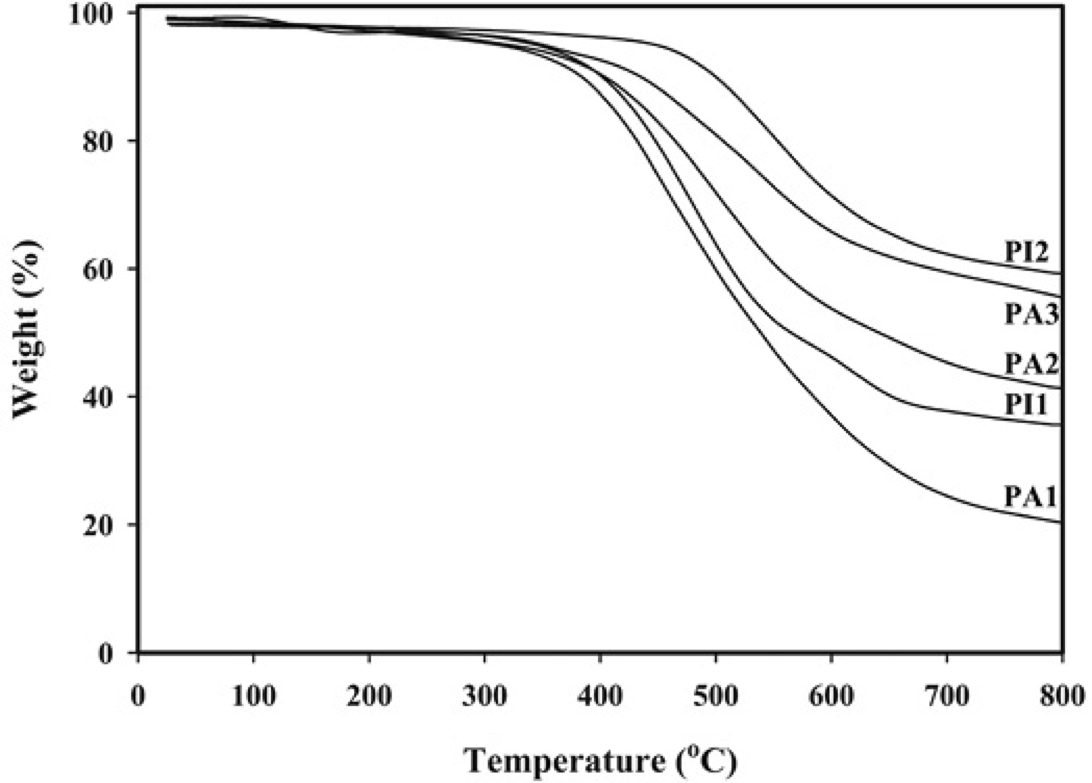
Thermal behavior of these PAs and PIs were evaluated by DSC and TGA measurements and the data extracted from the original curves were summarized in Table 3. DSC was used to determine the glass transition temperature (Tg) values of the polymers with a heating rate of 10 °C/min in N2 and the curves were shown in Fig. 8. The absence of the melting peak in the DSC thermograms supported the generally amorphous nature of these PAs and PIs. Synthesized PAs showed Tg values in the range of 220-286 °C, as listed in Table 3. Molecular packing state and chain rigidity are two main factors affecting Tg values. Although the increased rotational barrier caused by bulky aromatic pendent groups of diamine in these polymers and by additional -CF3 substituents enhanced Tg values, but the ether linkage inserted in the main chains of these polymers lead to lowering of Tg as well as significant improvement in solubility and processability. Moreover, the Tg values of these PAs depend on the stiffness of dicarboxylic acid component in the polymer chain. Therefore the aliphatic PA (PA1) showed lower Tg values in comparison with the aromatic PAs (PA2, PA3). The Tg values of PIs were 251 and 307 °C for PI1 and PI2, respectively. As the same as PAs, the Tg values decreased with increasing flexibility of the PIs backbones related to the applied structure of dianhydride from PI2 based on rigid phenyl groups dianhydride (PMDA) to PI1 based on bicyclic aliphatic dianhydride (BCDA). Thermal stability of polymers was evaluated by TGA in nitrogen atmosphere at a heating rate of 10 °C/min. The TGA curves are shown in Fig. 9. The temperatures of 5% weight loss (T5), the temperatures of 10% weight loss (T10) and the weight of polymer remained at 800 °C (Char yield) were determined from original TGA thermograms and tabulated in Table 3. Also, the results indicated PIs containing -CF3 groups showed lower thermal stability. Char yield can be used as criteria for evaluating limiting oxygen index (LOI) of the polymers in accordance with Van Krevelen and Hoftyzer equation [45] LOI= 17.5 + 0.4 CR where CR= char yield. For all the polymers LOI values calculated based on their char yield at 800 °C. In general, when LOI of a polymer is higher than 26 % it is considered to be self-extinguishable. According to Table 3, it is clear that aromatic PAs and PIs have better thermal stability and higher LOI as compared to the aliphatic PIs, because the aliphatic groups in the backbone of PA1 and PI1 increases the flexibility of the polymer, thereby decreasing the thermal stability. Therefore, these polymers possess excellent thermal stability because of their aromaticity, the strong electron negativity of the F atom and the high polarity of the CF3 group due to the high C-F bond strength and are candidates for applications in processable thermostable high-performance devices.
Table 3 Thermal characteristic data of PAs and PIs
Tg: glass transition temperature; T5: temperature for 5% weight loss; T10: temperature for 10% weight loss; Char yield: weight of polymer remained at 800 °C.
aLimiting oxygen index percent evaluating at char yield 800 ºC.

Adsorption of Heavy Metal Ions
In these experiments, the presence of imidazole rings, amide and ether linkages in the backbones of PAs and PIs can act as proper hosts for the adsorption and formation of complex with the target metal ions. Therefore, 5 mg of PA3 and PI2 were added to 25 mL of each metal ion solution, separately with initial metal ion concentration of 10 mg L-1 and stirred for 3 days at room temperature. After centrifuge separation, the amount of adsorbed ions was calculated using equations described in section 2 and the respective adsorption data, as summarized in Table 4, showed order of Cr3+ > Co2+ > Cd2+ > Hg2+ > Pb2+. As can be discussed from Table 4, the adsorption arrangement of metal ions has a reverse relation with radius of metal ions. In other words, polymers had the lower ability to adsorb larger metal ions due to the free space between the polymer chains which can allocate a place for the metal ions. As expected the adsorption values for PAs were higher than PIs due to existence of more hydrophilic amide linkages in PAs compare to PIs.
Conclusions
New diamine monomer was synthesized and reacted with various dicarboxylic acids and tetracarboxylic dianhydrides for the preparation of novel PAs and PIs, respectively. The results presented here demonstrate that different functional groups such as ether linkage and bulky groups such as CF3, phenyl groups and substituted imidazole along their backbones not only improved the solubility of the polymers in organic solvents while maintaining thermal stability with 10% weight loss temperatures at 383-512 °C. The results also demonstrated that presence of non-bonding electrons in ether, amide and imidazole linkages acted as hydrophilic chelating agents for heavy metal ions in the order of Cr3+ > Co2+ > Cd2+ > Hg2+ > Pb2+.
Materials
All materials and solvents were purchased either from Merck or Fluka Co (Germany). Ammonium acetate, acetic anhydride, hydrazine monohydrate, 10% palladium on activated carbon, bicyclo [2.2.2] oct-7-ene-2,3,5,6-tetracarboxylic dianhydride (BCDA), pyromellitic dianhydride (PMDA), reagent grade diacids such as terephthalic acid, pyridine-2,6-dicarboxylic acid, and sebacic acid were used as received. N-methyl-2-pyrrolidone (NMP), N,N-dimethylacetamide (DMAc) and pyridine (Py) were purified by distillation under reduced pressure over calcium hydride and stored over 4 A° molecular sieves. Tetrahydrofuran (THF) and toluene were dried by sodium wire before use. K2CO3 was dried for 24 h at 120 °C under vacuum. Co2+, Cr3+, Cd2+, Hg2+ and Pb2+ as their chloride and nitrate salts were used for the preparation of standard solutions. Distilled water (<1 microsiemens [μS]) was used for dilution of the stock solutions. All other reagents and solvents were used as received from commercial sources.
Measurements
Proton and carbon nuclear magnetic resonance (1H NMR) spectra were recorded on a 400 MHz Bruker Avance DRX (Germany) instrument using DMSO-d6 as solvent and tetramethyl silane as an internal standard. Proton resonances are designated as singlet (s), doublet (d), and multiplet (m). FT-IR spectra were recorded using a Bruker Tensor 27 spectrometer on KBr pellets over the range of 400-4000 cm-1. Elemental analyses performed by a CHN-600 Leco elemental analyzer. Melting point (uncorrected) was measured with a Barnstead Electrothermal engineering LTD 9200 apparatus. Inherent viscosities (at a concentration of 0.5 g/dL) were measured with an Ubbelohde suspended-level viscometer at 25 °C using NMP as solvent. Quantitative solubility was determined using 0.05 g of the polymer in 0.5 mL of solvent. Thermogravimetric analysis (TGA) was performed with the DuPont Instruments (TGA 951) analyzer well equipped with a PC at a heating rate of 10 °C/min under nitrogen atmosphere (20 cm3/min) and in the temperature range of 30-800 °C. Differential scanning calorimeter (DSC) was recorded on a Perkin Elmer pyris 6 DSC under nitrogen atmosphere (20 cm3/min) at a heating rate of 10 °C/min. Glass-transition temperatures (Tg) values were read at the middle of the transition in heat capacity and were taken from the second heating scan after cooling from 350 °C at a cooling rate of 20 °Cmin-1. Ultraviolet-visible and fluorescence emission spectra were recorded on a Cecil 5503 (Cecil Instruments, Cambridge, UK) and Perkin-Elmer LS-3B spectrophotometers (Norwalk, CT, USA) (slit width= 2 nm), respectively, using a dilute polymer solution (0.20 g/dL) in DMSO.
Adsorption Studies
For solid-liquid extraction of Co2+, Cr3+, Cd2+, Hg2+ and Pb2+, 5 mg of the appropriate polymer powder was shaken with 25 mL of aqueous solution of the metal salt with initial concentration of 10 mg L-1 for 3 days at 25 °C (pH 7-8). After centrifugal separation, the metal ion concentration in the supernatant was determined by AAS. The removal percentage (R%) of each metal ion and the adsorption capacity that represents the amount of adsorbed ions (mg g-1) were calculated using the following equations, respectively:
where C0 and CA are the concentrations of metal ions in the initial solution and in the aqueous phase after adsorption, respectively (mg L-1), Qt is the amount of metal ions adsorbed into the unit of the composites (mg g-1), V is the volume of the aqueous phase (mL) and W is the weight of the polymer (g).
Monomer Synthesis
Synthesis procedures of the target diamine are described below and the steps are shown in Scheme 1.
Synthesis of 2,2’-(6,6’)-(4,4-’(2-(4-triflouro methyl) phenyl)-1H-imidazole-4,5-dyl)bis (4,1-phenylen))bis(oxy)bis(3-nitro-1,6-phenylen))bis(4,5-diphenyl-1H-imidazole) (III)
Compounds (I) and (II) were synthesized according to the procedures given in the previous papers [19, 25]. Compound III was synthesized according to the following procedure.
Into a 100 mL two-necked round-bottomed flask equipped with a magnetic stirrer bar and a reflux condenser, 1 g (1 mol) compound I and 1.9 g (2 mol) compound II were dissolved in 20 mL N,N-dimethylacetamide, and 2.76 g (2.6 mol) potassium carbonate was added to the solution. After 30 min stirring at room temperature, the mixture was heated at 110 °C for 24 h. Then, the mixture was poured into 100 mL water and the precipitate was collected by filtration and dried in a vacuum oven at 80 °C. Yield: 2.2 g (81%), and mp: 327-334 °C. FT-IR (KBr disk) at cm-1: 3041 (aromatic C-H); 1520, 1355 (NO2); 1498 (C=C); 1289 (C-O-C), 698 (C-F). 1H NMR (DMSO-d6, δ in ppm): 6.09 (s, 2H, Ar-H), 7.17-7.36 (m, 22H, Ar-H), 7.44 (d, 2H, Ar-H, J= 7.6 Hz), 7.64 (d, 4H, Ar-H, J= 8.4 Hz), 7.77 (d, 4H, Ar-H, J= 7.2 Hz), 7.95 (d, 2H, Ar-H, J= 13.6 Hz), 11.12 (s, 2H, N-H imidazole), 13.57 (s, 1H, N-H imidazole). 13C NMR (DMSO-d6, δ in ppm): 120.36, 126.71, 126.78, 127.63, 127.90, 128.06, 128.59, 128.69, 129.04, 129.78, 131.10, 131.38, 133.55, 135.03, 136.67, 139.80, 167.43. Elemental analysis calculated for C64H41N8O6F3: C, 71.50 %; H, 3.84 %; N, 10.42%. Found: C, 71.44 %; H, 3.86%; N, 10.47%.
Synthesis of 4,4’-(4,4’-(2-(4-triflouro methyl) phenyl)-1H-imidazole-4,5-dyl) bis (1,4phenylen)) bis (oxy) bis (3-(4,5-diphenyl-1H-imidazole-2-yl) aniline) (IV)
Into a 100 mL three-necked round-bottomed flask equipped with a dropping funnel, a reflux condenser and a magnetic stirrer bar, a mixture of 1 g (0.001 mol) compound III and 0.1 g of 10% Pd/C were dispersed in 50 mL ethanol. The suspension was heated to reflux at 70 °C and 1.5 mL hydrazine monohydrate was added slowly to the mixture through dropping funnel. After a further 5 h reflux, the temperature was increased up to 90 °C and the solution was filtered hot to remove Pd/C. The filtrate was cooled to give a brownish precipitate which was recrystallized in methanol, and dried in a vacuum oven at 80 °C. Yield: 0.75 g (79%) and mp: 223-227 °C. FT-IR (KBr disk) at cm-1: 3404, 3281 (N-H), 3060 (aromatic C-H), 1500 (C=C), 1383 (C-N), 1244 (C-O-C), 775 (C-F). 1H NMR (DMSO-d6, δ in ppm): 4.41 (s, 4H, NH2), 6.17 (dd, 2H, Ar-H), 6.47-6.70 (m, 4H, Ar-H), 7.12-7.30 (m, 20H, Ar-H), 7.42-7.84 (m, 8H, Ar-H), 8.00 (d, 2H, Ar-H, J= 7.2 Hz), 8.27 (d, 2H, Ar-H, J= 7.6 Hz), 10.55 (s, 2H, N-H imidazole), 13.13 (s, 1H, N-H imidazole). 13C NMR (DMSO-d6, δ in ppm): 114.34, 115.43, 117.11, 117.24, 117.72, 125.38, 126.60, 126.72, 127.18, 128.02, 128.55, 128.80, 129.05, 130.44, 131.50, 131.64, 134.10, 134.28, 139.40, 140.47, 145.67, 148.16, 167.40. Elemental analysis calculated for C64H45N8O2F3: C, 75.73%; H, 4.47%; N, 11.04%. Found: C, 75.81%; H, 4.42%; N, 11.10%.
Polyamide Synthesis
General procedure for the preparation of PAs is illustrated in Scheme 2. In a 50 mL two-necked round-bottomed flask equipped with a reflux condenser, a nitrogen gas inlet tube, and a magnetic stirrer bar, a mixture of 1.5 mL TPP, 2 mL Py, 0.2 g LiCl, 0.4 g CaCl2, 5.0 mL NMP, 0.2 g (0.001 mol) target diamine (compound IV), and 0.001 mol of a dicarboxylic acid such as sebacic acid, 2,6-pyridine dicarboxylic acid and terephthalic acid was refluxed under a stream of N2 at 130 °C for 12 h to produce PA1, PA2 and PA3, respectively. After cooling the flask to room temperature, the polymer solution was poured into a 150 mL methanol. The formed precipitate was collected by filtration and washed thoroughly with hot water. The obtained PAs were extracted in refluxing methanol to remove low molecular weight oligomers and dried at 80 °C in a vacuum oven for 24 h.
PA1: This polymer was obtained from diamine (IV) and sebacic acid. Yield 88%. FT-IR (KBr disk, cm-1): 3425 (N-H imidazole), 3276 (N-H amide), 3059 (C-H aromatic), 2926 (C-H aliphatic), 1656 (C=O amide), 1596 (C=N), 1481 (C=C) and 1156 (C-O-C), 764 (C-F). 1H NMR (DMSO-d6, δ in ppm): 1.67 (s, 8H, C-H), 2.08 (s, 4H, C-H), 2.39 (t, 4H, C-H), 6.74-8.11 (m, 38H, Ar-H), 10.16 (s, 1H, N-H amide), 10.27 (s, 1H, N-H amide), 11.30 (s, 2H, N-H imidazole ring), 12.80 (s, 1H, N-H imidazole ring). Elemental analysis calculated for (C74H63N6O4F3)n: C, 75.25%; H, 5.00%; N, 9.49%. Found: C, 75.18%; H, 5.08%; N, 9.45%.
PA2: This polymer was obtained from diamine (IV) and terephthalic acid. Yield 85%. FT-IR (KBr disk, cm-1): 3424 (N-H imidazole), 3273 (N-H amide), 3061 (C-H aromatic),1657 (C=O amide), 1603 (C=N), 1483 (C=C) and 1249 (C-O-C), 768 (C-F).1H NMR (DMSO-d6, δ in ppm): 7.27-8.69 (m, 42H, Ar-H), 10.69 (s, 1H, N-H amide), 10.80 (s, 1H, N-H amide), 11.30 (s, 2H, N-H imidazole ring), 13.04 (s, 1H, N-H imidazole ring). Elemental analysis calculated for (C72H47N8O4F3)n: C, 75.52%;H, 4.10%; N, 9.79%. Found: C, 75.41%; H, 4.23%; N, 9.75%.
PA3: This polymer was obtained from diamine (IV) and 2, 6-pyridine dicarboxylic acid. Yield 89%. FT-IR (KBr disk, cm-1): 3425 (N-H imidazole), 3282 (N-H amide), 3047 (C-H aromatic), 1656 (C=O amide), 1583 (C=N), 1480 (C=C) and 1250 (C-O-C), 769 (C-F).1H NMR (DMSO-d6, δ in ppm): 6.84-8.63 (m, 41H, Ar-H), 11.01 (s, 1H, N-H amide), 11.23 (s, 1H, N-H amide), 11.41 (s, 2H, N-H imidazole ring), 13.03 (s, 1H, N-H imidazole ring). Elemental analysis calculated for (C71H46N9O4F3)n: C, 74.41%; H, 4.01%; N, 11.00%. Found: C, 74.53%; H, 3.96%; N, 10.95%.
Polyimides Synthesis
The commercially available cyclic and aromatic dianhydrides, BCDA and PMDA, were reacted with the target diamine (IV) for the preparation of PI-1 and PI-2, respectively. General procedure is illustrated in Scheme 2. A 50 mL two-necked round-bottomed flask equipped with a reflux condenser, a magnetic stirrer bar, and a nitrogen gas inlet tube was charged with 0.2 g (0.001 mol) IV and 5 mL dry NMP. The mixture was stirred at room temperature for 0.5 h. After adding 0.001 mol BCDA or PMDA, the mixture was stirred at room temperature for 24 h to form a viscous solution of poly (amic acid) (PAA) precursor. Then 3 mL acetic anhydride/pyridine (6/4, v/v) solution was added to the PAA solution and the mixture was stirred at room temperature for 0.5 h, during this time PAA is converted to polyimide through chemical imidization process. The mixture was stirred at 130 °C for 10 h to yield a homogeneous solution. The polymer solution was poured slowly into methanol to form a precipitate. The precipitate was collected by filtration, washed thoroughly with hot water. The obtained PIs were extracted in refluxing methanol to remove low molecular weight oligomers and dried at 80 °C in a vacuum oven for 24 h.
PI1: This polymer was obtained from diamine (IV) and bicyclo [2.2.2] oct-7-ene-2,3,5,6-tetracarboxylic dianhydride. Yield= 91%. FT-IR (KBr, cm-1): 3424 (N-H imidazole), 3060 (C-H stretching aromatic), 2967 (C-H stretching Aliphatic), 1773 and 1724 (imide carbonyl asymmetric and symmetric stretching), 1481 (C=N stretching), 1359 (C-N stretching), 1112 (C-O-C), 1080 and 697 (imide ring deformation), and 768 (C-F). 1H NMR (DMSO-d6, δ in ppm) δ: 2.68-2.85 (m, 2H, C-H), 3.05-3.36 (m, 4H, C-H), 5.57 (m, 2H, C-H), 7.06-8.61 (m, 38H, Ar-H), 12.26 (s, 2H, N-H imidazole ring), 12.42 (s, 1H, N-H imidazole ring). Elemental analysis calculated for [C76H49N8O6F3]n: C, 74.38%; H, 3.99%; N, 9.13%. Found: C, 74.32%; H, 3.92%; N, 9.22%.
PI2: This polymer was obtained from diamine (IV) and pyromellitic dianhydride. Yield = 86%. FT-IR (KBr, cm-1): 3441 (N-H imidazole), 3059 (C-H stretching Aromatic), 1779 and 1712 (imide carbonyl asymmetric and symmetric stretching), 1485 (C=N stretching), 1297 (C-N stretching), 1236 (C-O-C), 1173 and 698 (imide ring deformation), 770 (C-F). 1H NMR (DMSO-d6, δ in ppm) δ: 7.16-8.23 (m, 40H, Ar-H), 12.17 (s, 2H, N-H imidazole ring), 12.34 (s, 1H, N-H imidazole ring). Elemental analysis calculated for [C74H43N8O6F3]n: C, 74.24%; H, 3.59%; N, 9.36%. Found: C, 74.29%; H, 4.66%; N, 9.28%.

