











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Synthesis of heterocyclic compounds has emerged as a powerful technique for generating new molecules useful for drug discovery [1]. Heterocyclic compounds containing nitrogen and oxygen plays important role in agrochemical and pharmaceuticals. Benzofuran derivatives posses a wide range of biological activities such as antimicrobial [2], antitumour [3], anti-inflammatory [4], antidepressant [5], analgesic [6] and hypoglycaemic [7] activities. Benzobromarone [8], a uricosuric agent and 5-methyl-3-p-toluoyl-2[4-(3-diethylaminopropoxy) phenyl] benzofuran [9], which is a β-amyloid aggregation inhibitor are the examples of pharmacologically active benzofuran derivatives (Fig. 1).

Pyrazole, a five-membered aromatic nitrogen heterocycle forms a part of the structure of several natural products and drugs. Pyrazole derivatives are known to possess antimicrobial[10], antitumor [11-12], antihypertensive [13], antidepressant [14], insecticidal [15], antifungal [16], 5α-reductase-inhibitory [17], antiproliferative [18], antiparasitic [19], herbicidal [20], anti-inflammatory [21-22], antiprotozoal [23], analgesic[22] and androgen receptor modulatory [24] activities. The pyrazole ring is present as the core in a variety of leading drugs such as lonazolac [25] and rimonabant [26] (Fig. 1). Thiophene structure can be found in certain natural products and is also incorporated in several pharmacologically active compounds. In medicinal chemistry, thiophene derivatives have been very well known for their therapeutic applications e.g. tienilic acid, a loop diuretic drug [27] and 4-hydrazino-2-mehylthio-5-ethyl-6-methylthieno[2,3-d]pyrimidine, an antimicrobial agent [28] (Fig. 1).
Aryl-aryl and aryl-vinyl bond formation is very important in organic synthesis and has a wide range of applications of industrial interest, including the synthesis of pharmaceuticals, polymers, herbicides and other materials [29]. Some natural biaryl derivatives can be used as drugs and an example of this is mastigophorene A (Fig 1) [30]. The palladium catalysed Suzuki cross coupling between organoboron compounds and organic halides leads to the formation of carbon-carbon bond, was first reported by Suzuki and Miyayura and it was a revolution in organic couplings [31]. For synthesis of biaryl compounds, palladium-catalyzed Suzuki-Miyaura coupling is the most attractive method [32]. Pd(Ph3)4, one of the most suitable catalyst for Suzuki-Miyaura coupling because of the functional-group tolerance, and low sensitivity towards water and air so that reactions can takes place even in aqueous medium [33].
Nowadays, organic synthesis in aqueous phase or including water and water/organic mixture as a solvent has been received great attention in the field of organic chemistry. Also, it is familiar that microwave assisted organic synthesis (MAOS) is environmental friendly and allows simple separation and catalyst recycling [34]. So, by the microwave assisted synthesis using water as a solvent, one can easily achieve the purpose of green chemistry. In view of the potential bioactivity of benzofuran, biaryl and pyrazole moieties, we have taken up the synthesis of some new benzofuranpyrazolebiaryl derivatives using Suzuki coupling by the combination of two prominent green chemistry principles, namely microwave assisted synthesis and aqueous phase reactions. We have taken up the synthesis by microwave as well as conventional methods. All the synthesized compounds have tested in vitro for their antibacterial and antifungal activities.
Results and Discussion
The synthetic route to compounds 7a-j is presented in Scheme 1. The condensation of 2-hydroxy acetophenone (1) with 1-phenyl-3-(thiophen-2-yl)-1H-pyrazole-4-carboxaldehyde [35] (2) in the presence of alkali under microwave irradiation [36] gave (E)-1-(2-hydroxyphenyl)-3-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4- yl)prop-2-en-1-one [37] (3) in 95% yield. In the MS of 3 the base peak appeared at 551 corresponding to (M+H)+. Subsequently, this chalcone (3) on reaction with 2-bromo-1-(4-bromophenyl)ethanone (4) in presence of potassium carbonate, yielded new benzofuran derivative, (E)-(4-bromophenyl)(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanone (5). Compound 5 was characterized as (E)-(4-bromophenyl)(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanone. In the IR spectrum of 5, a peaks at 825, 1216, 1740 cm-1 confirms the presence of C-Br, C-O-C and carbonyl groups respectively. In 1H NMR spectrum of 5 showed doublet at δ 7.81 ppm corresponding to two aromatic protons which are ortho to carbonyl group. In 13C NMR the carbonyl peak is observed at δ 185.7 ppm.

Compound 5 on Suzuki coupling with different aryl boronic acids (6a-j) yielded the target molecules (E)-(4-(aryl)phenyl)(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanones (7a-j). Synthesis of compounds 7a-j was performed by conventional and microwave irradiation techniques [38] (Table 2). Based on the literature precedence [39] we started our initial studies for the Suzuki coupling reaction with Na2CO3 as a base and Pd(PPh3)4 as a catalyst. Catalyst loading screening studies tells that increasing the amount of Pd(PPh3)4 helping for the good yields (Table 1). Finally we arrived, 15 mol % of Pd(PPh3)4 as a optimal condition where we obtained 80% of sole product (7a). Compound 7a was characterized as (E)-[1,1’-biphenyl]-4-yl(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanone. All peaks in 1H NMR spectrum of 7a support the formation of product and in 13C NMR of 7a the carbonyl peak is observed at δ 185.3 ppm. In the MS of 7a the appearance of base peak at 548 corresponding to (M+H)+ confirms the formation of Suzuki coupled product.

Antibacterial activity
The synthesized novel compounds 5 and 7a-j were screened for their antibacterial activity against different types of bacterial strains, they are Gram positive bacterial strains of Bacillus subtillis and Staphylococcus aeureus, Gram negative bacterial strains of Klebsiella pneumoniae and Escherichia coli at a concentration of 10 µg/mL and 20 µg/mL (Fig. 2). Some of the synthesized compounds shown high activity and some shown moderate activity compared to standard drug Gatifloxacin at a concentration of 10 µg/mL and 20 µg/mL. The antibacterial activity of compound 7e (Ar= 4-carboxyphenyl), 7g (Ar= 4-formylphenyl) and 7b (Ar=4-fluorophenyl) shown high zone of inhibition against Staphylococcus aeureus, Bacillus subtilis, Escherichia coli and Klebsiella pneumoniae compared to the standard drug at a concentration of 10 µg/mL and 20 µg/mL. Whereas the compounds 7c (Ar= 4- chlorophenyl) and 7d (Ar=2,4-dichlorophenyl) were almost equal to the standard drug at a concentration of 10 µg/mL and 20 µg/mL.

Antifungal activity
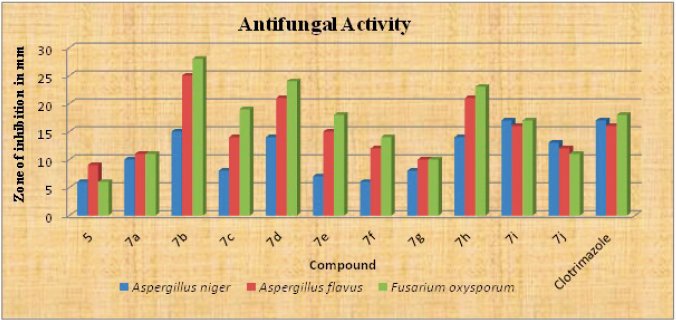
The antifungal activity of synthesized compounds 5 and 7a-j were tested against three pathogenic fungi, namely Fusarium oxysporum, Aspergillus nigerzeae, and Aspergilus flavus, by the poison plate technique at a concentration of 100 µg/mL (Fig. 3). Some of the synthesized compounds shown moderate to high antifungal activity compared to standard drug clotrimazole at a concentration of 100 µg/mL. The antifungal activity of compounds 7b (Ar= 4-fluorophenyl), 7d (Ar= 2,4- dichlorophenyl) and 7h (Ar=3-formyl) shown high zone of inhibition against Aspergilus flavus and Fusarium oxysporum compared to the standard drug. Compounds 7c (Ar= 4-chlorophenyl), 7e (Ar= 4-carboxyphenyl) shown high zone of inhibition against Fusarium oxysporum compared to the standard drug. The compound 7i (Ar= 3-fluoro-5-methylphenyl) shown zone of inhibition against tested fungi were almost equal to the standard drug. Rest of the compounds shown moderate activity against pathogenic fungi compared to standard.
Conclusions
A series of some new (E)-(4-(aryl)phenyl)(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanones (7a-j) derivatives were prepared by traditional Claisen-Schmidt and Suzuki cross-coupling reactions under conventional and microwave irradiation methods. All final compounds were investigated for their in vitro antimicrobial activity. Some of the compounds show better antimicrobial activity against selected microorganisms compared with the reference drugs. Thus, the above compounds can be considered as lead compounds for further development of more potent antimicrobial agents.
EXPERIMENTAL
General
Stuart SMP30 apparatus is used to determine melting points. Purity of the compounds was checked by TLC on silica gel 60 F254 (Merck). All the microwave irradiation experiments were performed in a CEM Discover microwave system and reaction temperatures were monitored by an equipped IR sensor. 1H NMR and 13C NMR spectra were recorded on Bruker Avance II 400 spectrometer using TMS as an internal standard. IR spectra were recorded in KBr on a Shimadzu FTIR 8400S spectrophotometer. Mass spectra were recorded on a Shimadzu GCMS-QP 1000 mass spectrometer. The elemental analysis was carried out on a Vario-11CHN analyzer.
Procedure for the synthesis of compound (E)-1-(2-hydroxyphenyl)-3-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)prop-2-en-1-one (3):
A mixture of 2-hydroxyacetophenone (1) (240 mg, 2 mmol), 1-phenyl-3-(thiophen-2-yl)-1H-pyrazole-4-carboxaldehyde (2) (508 mg, 2 mmol) in ethanol (2 mL) and NaOH (200 mg, 5 mmol) was introduced into CEM discover microwave reaction vessel equipped with a magnetic stirrer. The vessel was sealed and then placed into the microwave cavity. Initial microwave irradiation of 100 W was used, the temperature being ramped from room temperature to the desired 80 ºC temperature. Progress of the reaction was monitored by TLC. After completion of the reaction (10 min) it was diluted with cold water and acidified with diluted HCl, the precipitate formed was filtered, dried and recrystallized from ethanol to afford pure chalcone (3) (Yield: 95%).
A yellowish solid: mp 202-204 oC; IR (KBr) νmax 3416 (OH), 1681 (C=O), 1620 (C=CH) cm-1. 1H NMR (CDCl3, 400 MHz) δ 6.92-6.96 (m, 1H, Ar-H), 7.03 (d, J = 9.0 Hz, 1H, Ar-H), 7.18-7.20 (m, 1H, Ar-H), 7.35-7.56 (m, 7H, Ar-H, Hα), 7.80 (d, J = 7.7 Hz, 2H, Ar-H), 7.86 (d, J = 7.78 Hz, 1H, Ar-H), 8.14 (d, J = 15.3 Hz, 1H, Hβ), 8.38 (s, 1H, pyrazole-H), 12.90 (s, 1H, OH); 13C NMR (CDCl3, 100 MHz) δ 110.3, 117.9, 118.9, 119.4, 120.6, 121.2, 126.9, 127.0, 127.3, 127.6, 127.9, 129.6, 131.6, 133.5, 136.5, 138.7, 139.0, 148.3, 162.5, 192.2; Anal. Calcd for C22H16N2O2S: C, 70.95; H, 4.33; N, 7.52. Found: C, 70.90; H, 4.29; N, 7.46.
Procedure for the synthesis of compound (E)-(4-bromophenyl)(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanone (5)
Conventional method: A mixture of (E)-1-(2-hydroxyphenyl)-3-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)prop-2-en-1-one (3) (372 mg, 1 mmol) and 2-bromo-1-(4-bromophenyl)ethanone (4) (276 mg, 1 mmol), 5 g of anhydrous K2CO3 and acetone (15 mL) was taken in 100 mL round bottomed flask and it was refluxed for 4 h. After completion of reaction as followed by the TLC examination, cold water was added. The solid separated was filtered and washed with water and extracted with hot 5% NaOH solution. The alkaline solution was cooled to 0- 5 ºC and neutralized with diluted HCl. The precipitated solid was filtered, washed with cold water and dried. The crude product was purified by column chromatography on silica gel using hexane:ethyl acetate (7:3 v/v) as eluent and further recrystallized from methanol to afford the desired product 5 (Yield 75%).
Microwave irradiation method: A mixture of (E)-1-(2-hydroxyphenyl)-3-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)prop-2-en-1-one (3) (372 mg, 1 mmol) and 2-bromo-1-(4-bromophenyl)ethanone (4) (276 mg, 1 mmol) was dissolved in minimum quantity of ethanol and adsorbed by 5 g of anhydrous K2CO3. This was introduced into CEM discover microwave reaction vessel equipped with a magnetic stirrer. The vessel was sealed and then placed into the microwave cavity. Initial microwave irradiation of 300 W was used, the temperature being ramped from room temperature to 120 ºC temperature. Once this was reached, the reaction mixture was heated at this temperature for 6 min. After completion of reaction as followed by the TLC examination, cold water was added to. The solid separated was filtered and washed with water and extracted with hot 5% NaOH solution. The alkaline solution was cooled to 0- 5 ºC and neutralized with diluted HCl. The precipitated solid was filtered, washed with cold water and dried. The crude product was purified by column chromatography on silica gel using hexane:ethyl acetate (7:3 v/v) as eluent and further recrystallized from methanol to afford the desired product 5 (Yield: 88%)
A pale yellow solid: mp 232-234 oC; IR (KBr) νmax 1740 (C=O), 1602 (C=CH), 1216 (C-O-C), 825 (C-Br) cm-1. 1H NMR (CDCl3, 400 MHz) δ 7.21-7.23 (m, 1H, Ar-H), 7.32-7.36 (m, 1H, Ar-H), 7.45-7.57 (m, 7H, Ar-H, H-1’’), 7.63-7.67 (m, 3H, Ar-H), 7.81 (d, J= 7.5 Hz, 2H, Ar-H), 7.89 (d, J= 16.8 Hz, 1H, H-2’’), 8.07-8.09 (m, 2H, Ar-H), 8.29 (d, J= 1.7 Hz, 1H, Ar-H), 8.33 (s, 1H, pyrazole-H); 13C NMR (CDCl3, 100 MHz) δ 113.7, 119.2, 119.6, 119.9, 122.7, 124.9, 125.3, 125.4, 126.1, 126.6, 126.9, 127.1, 127.7, 128.4, 128.7, 129.5, 129.8, 129.9, 131.3, 134.7, 137.6, 139.5, 146.4, 148.6, 153.0, 185.7; ESI MS m/z (rel int): 551 (M+H)+(100). Anal. Calcd for C30H19BrN2O2S: C, 65.34; H, 3.47; N, 5.08. Found: C, 65.31; H, 3.44; N, 5.02.
General procedure for the synthesis of compounds (E)-(4-(aryl)phenyl)(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanones (7a-j):
Conventional heating method:
To a round bottomed flask was added (E)-(4-bromophenyl)(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanone (5) (550 mg, 1 mmol), Pd(PPh3)4 (0.15 mmol), arylboronic acid (1.30 mmol) (6a-j), Na2CO3 (3.00 mmol) and water (20 mL) sequentially. The mixture was degassed with nitrogen over 15 min. Then, the reaction mixture was stirred at 80°C for 5-7 h (Table 2) under nitrogen atmosphere. After cooled to room temperature, the mixture was diluted with EtOAc (15 mL) and washed with H2O and brine solution. The organic layer was washed with 10 mL of water and it was dried over anhydrous magnesium sulphate. The solvent was evaporated and the residue was purified by using silica gel column chromatography (1:3 v/v ethylacetate: hexane) to afford pure products (7a-j).
Microwave irradiation method: A degassed mixture of (E)-(4-bromophenyl)(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanone (5) (550 mg, 1 mmol), Pd(PPh3)4 (0.15 mmol), arylboronic acid (1.30 mmol) (6a-j), Na2CO3 (3.00 mmol) and water (5 mL) was introduced into CEM discover microwave reaction vessel equipped with a magnetic stirrer. The vessel was sealed and then placed into the microwave cavity. Initial microwave irradiation of 180 W was used, the temperature being ramped from room temperature to the desired 80 ºC temperature. Once this was reached, the reaction mixture was heated at this temperature for appropriate time as shown in Table 2. The reaction mixture was stirred continuously during the reaction. After cooled to room temperature, the mixture was diluted with EtOAc and washed with H2O and brine solution. The organic layer was washed with 10 mL of water and it was dried over anhydrous magnesium sulphate. The solvent was evaporated and the residue was purified by using silica gel column chromatography (1:3 v/v ethylacetate: hexane) to afford pure products (7a-j).
(E)-[1,1’-biphenyl]-4-yl(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanone (7a). A pale yellow solid: mp 225-227 oC; IR (KBr) νmax 1739 (C=O), 1598 (C=CH), 1216 (C-O-C) cm-1. 1H NMR (CDCl3, 400 MHz) δ 7.19-7.21 (m, 1H, Ar-H), 7.30-7.34 (m, 1H, Ar-H), 7.41-7.53 (m, 8H, Ar-H), 7.54-7.58 (m, 1H, Ar-H), 7.63 (d, J= 7.5 Hz, 1H, Ar-H), 7.68-7.81 (m, 7H, Ar-H, H-1’’), 8.00 (d,J = 16.5 Hz, 1H, H-2’’), 8.15 (d, J= 8.0 Hz, 1H, Ar-H), 8.21 (d, J = 8.7 Hz, 2H, Ar-H), 8.33 (s, 1H, pyrazole-H); 13C NMR (CDCl3, 100 MHz) δ 112.7, 119.1, 120.2, 120.4, 123.1, 124.2, 124.8, 124.9, 125.9, 126.0, 126.1, 126.8, 127.0, 127.2, 127.3, 127.7, 128.2, 128.3, 129.0, 129.4, 130.5, 134.9, 136.6, 139.5, 139.9, 145.4, 146.3, 147.9, 154.8, 185.3; ESI MS m/z (rel int): 549 (M+H)+(100). Anal. Calcd for C36H24N2O2S: C, 78.81; H, 4.41; N, 5.11. Found: C, 78.75; H, 4.36; N, 5.07.
(E)-(4’-fluoro-[1,1’-biphenyl]-4-yl)(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanone (7b). A pale yellow solid: mp 242-244 oC; IR (KBr) νmax 1741 (C=O), 1599 (C=CH), 1235 (C-O-C), 696 (C-F) cm-1. 1H NMR (CDCl3, 400 MHz) δ 7.15-7.21 (m, 3H, Ar-H), 7.31-7.34 (m, 1H, Ar-H), 7.41-7.66 (m, 9H, Ar-H, H-1’’), 7.72-7.80 (m, 5H, Ar-H), 7.98 (d, J = 16.8 Hz, 1H, H-2’’), 8.15 (d, J = 8.0 Hz, 1H, Ar-H), 8.20 (d, J = 8.5 Hz, 2H, Ar-H), 8.33 (s, 1H, pyrazole-H); 13C NMR (CDCl3, 100 MHz) δ 113.7, 114.1, 117.3, 119.2, 119.5, 119.6, 119.9, 122.7, 124.9, 125.3, 125.4, 125.8, 126.1, 126.2, 126.4, 126.6, 126.9, 127.7, 128.4, 128.7, 129.5, 129.8, 129.9, 131.3, 132.9, 134.6, 137.6, 139.5, 146.5, 148.6 (J= 244), 153.3, 185.7; ESI MS m/z (rel int): 567 (M+H)+(100). Anal. Calcd for C36H23FN2O2S: C, 76.31; H, 4.09; N, 4.94. Found: C, 76.27; H, 4.03; N, 4.91.
(E)-(4’-chloro-[1,1’-biphenyl]-4-yl)(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanone (7c). A pale yellow solid: mp 232-234 oC; IR (KBr) νmax 1738 (C=O), 1586 (C=CH), 1230 (C-O-C), 744 (C-Cl) cm-1. 1H NMR (CDCl3, 400 MHz) δ 7.19-7.21 (m, 1H, Ar-H), 7.31-7.35 (m, 1H, Ar-H), 7.40-7.62 (m, 10H, Ar-H, H-1’’), 7.70-7.81 (m, 6H, Ar-H), 7.99 (d, J = 16.8 Hz, 1H, H-2’’), 8.14 (d, J = 8.0 Hz, 1H, Ar-H), 8.20 (d, J = 8.2 Hz, 2H, Ar-H), 8.35 (s, 1H, pyrazole-H); 13C NMR (CDCl3, 100 MHz) δ 112.2, 119.2, 120.2, 120.3, 124.3, 124.9, 125.2, 125.8, 125.9, 126.0, 126.1, 126.9, 127.0, 127.6, 127.7, 128.0, 128.5, 129.5, 130.6, 134.9, 135.5, 137.7, 139.5, 142.4, 142.9, 147.7, 154.8, 186.7; ESI MS m/z (rel int): 583 (M+H)+(100). Anal. Calcd for C36H23ClN2O2S: C, 74.15; H, 3.98; N, 4.80. Found: C, 74.11; H, 3.93; N, 4.76.
(E)-(2’,4’-dichloro-[1,1’-biphenyl]-4-yl)(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanone (7d). A pale yellow solid: mp 236-238 oC; IR (KBr) νmax 1740 (C=O), 1598 (C=CH), 1233 (C-O-C), 742 (C-Cl) cm-1. 1H NMR (CDCl3, 400 MHz) δ 7.18-7.20 (m, 1H, Ar-H), 7.31-7.60 (m, 10H, Ar-H, H-1’’), 7.68-7.82 (m, 6H, Ar-H), 7.95-8.00 (m, 3H, Ar-H, H-2’’), 8.13 (d, J= 8.0 Hz, 1H, Ar-H), 8.34 (s, 1H, pyrazole-H); 13C NMR (CDCl3, 100 MHz) δ 112.1, 119.4, 121.2, 123.3, 124.3, 124.8, 125.2, 125.8, 125.6, 126.0, 126.1, 126.9, 127.0, 127.6, 127.7, 128.0, 128.5, 129.5, 130.6, 134.9, 135.5, 137.7, 139.5, 142.4, 142.9, 147.3, 154.5, 186.5; ESI MS m/z (rel int): 617 (M+H)+(100). Anal. Calcd for C36 H22Cl2N2O2S: C, 70.02; H, 3.59; N, 4.54. Found: C, 69.97; H, 3.54; N, 4.51.
(E)-4’-(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-carbonyl)-[1,1’-biphenyl]-4-carboxylic acid (7e). A pale yellow solid: mp 248-250 oC; IR (KBr) νmax 1739 (C=O), 1598 (C=CH), 1216 (C-O-C) cm-1. 1H NMR (CDCl3, 400 MHz) δ 7.18-7.20 (m, 1H, Ar-H), 7.31-7.34 (m, 1H, Ar-H), 7.45-7.84 (m, 14H, Ar-H, H-1’’), 7.98-8.00 (m, 3H, Ar-H, H-2’’), 8.13 (d, J=8.0 Hz, 1H, Ar-H), 8.25 (d, J=8.5 Hz, 2H, Ar-H) 8.33 (s, 1H, pyrazole-H), 13.09 (s, 1H, -COOH); 13C NMR (CDCl3, 100 MHz) δ 112.6, 119.2, 120.1, 123.2, 124.3, 125.0, 125.3, 125.8, 126.0, 126.1, 126.9, 127.7, 127.8, 128.6, 129.5, 131.3, 131.6, 134.9, 136.6, 139.5, 146.3, 147.4, 154.8, 180.5, 187.3; ESI MS m/z (rel int): 593 (M+H)+(100). Anal. Calcd for C37H24N2O4S: C, 74.98; H, 4.08; N, 4.73. Found: C, 74.92; H, 4.04; N, 4.69.
(E)-(4’-methyl-[1,1’-biphenyl]-4-yl)(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanone (7f). A pale yellow solid: mp 212-214 oC; IR (KBr) νmax 1739 (C=O), 1598 (C=CH), 1216 (C-O-C) cm-1. 1H NMR (CDCl3, 400 MHz) δ 2.42 (s, 3H, Me-H), 7.18-7.20 (m, 1H, Ar-H), 7.33-7.40 (m, 3H, Ar-H), 7.42-7.63 (m, 9H, Ar-H, H-1’’), 7.70-7.79 (m, 5H, Ar-H), 7.96 (d, J = 8.0 Hz, 1H, Ar-H), 8.13-8.19 (m, 3H, Ar-H, H-2’’), 8.31 (s, 1H, pyrazole-H); 13C NMR (CDCl3, 100 MHz) δ 21.1, 112.7, 119.1, 120.2, 120.4, 123.1, 124.2, 124.7, 124.9, 125.9, 126.1, 126.0, 126.7, 126.8, 127.1, 127.7, 128.3, 129.4, 129.7, 130.5, 135.0, 136.4, 137.0, 138.2, 139.5, 145.4, 146.3, 148.0, 154.8, 185.3; ESI MS m/z (rel int): 563 (M+H)+(100). Anal. Calcd for C37H26N2O2S: C, 78.98; H, 4.66; N, 4.98. Found: C, 78.92; H, 4.62; N, 4.94.
(E)-4’-(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-carbonyl)-[1,1’-biphenyl]-4-carbaldehyde (7g). A pale yellow solid: mp 246-248 oC; IR (KBr) νmax 1740 (C=O), 1670 (C=O of aldehyde), 1602 (C=CH), 1216 (C-O-C) cm-1. 1H NMR (CDCl3, 400 MHz) δ 6.96 (d, J = 8.5 Hz, 1H, Ar-H), 7.19-7.35 (m, 2H, Ar-H), 7.44-8.02 (m, 16H, Ar-H, H-1’’, H-2’’), 8.15 (d, J = 7.7 Hz, 1H, Ar-H), 8.15 (d, J = 8.2 Hz, 1H, Ar-H), 8.34 (s, 1H, pyrazole-H); 13C NMR (CDCl3, 100 MHz) δ 112.7, 115.9, 119.2, 120.2, 120.3, 123.2, 124.3, 125.0, 125.2, 125.9, 126.0, 126.1, 126.9, 127.3, 127.7, 127.9, 128.5, 129.0, 129.1, 129.5, 130.3, 130.5, 130.6, 131.6, 135.8, 134.9, 137.7, 139.5, 154.8, 185.2, 191.8; ESI MS m/z (rel int): 577 (M+H)+(100). Anal. Calcd for C37H24N2O3S: C, 77.06; H, 4.19; N, 4.86. Found: C, 77.02; H, 4.14; N, 4.82.
(E)-4’-(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-carbonyl)-[1,1’-biphenyl]-3-carbaldehyde (7h). A pale yellow solid: mp 228-230 oC; IR (KBr) νmax 1740 (C=O), 1695 (C=O of aldehyde), 1602 (C=CH), 1231 (C-O-C) cm-1. 1H NMR (CDCl3, 400 MHz) δ 7.19-7.21 (m, 1H, Ar-H), 7.31-7.35 (m, 1H, Ar-H), 7.44-7.83 (m, 13H, Ar-H, H-1’’), 7.92-8.03 (m, 3H, Ar-H, H-2’’), 8.14-8.25 (m, 4H, Ar-H), 8.35 (s, 1H, pyrazole-H), 10.12 (s, 1H, -CHO); 13C NMR (CDCl3, 100 MHz) δ 112.7, 119.2, 120.2, 120.3, 123.1, 124.3, 124.9, 125.1, 125.9, 126.0, 126.1, 126.9, 127.0, 127.5, 127.7, 128.5, 129.5, 129.5, 129.7, 130.6, 133.1, 134.9, 137.0, 137.3, 139.5, 140.9, 143.7, 146.3, 147.8, 154.8, 185.2, 192.0; ESI MS m/z (rel int): 577 (M+H)+(100). Anal. Calcd for C37H24N2O3S: C, 77.06; H, 4.19; N, 4.86. Found: C, 77.02; H, 4.14; N, 4.82.
(E-(3’-fluoro-5’-methyl-[1,1’-biphenyl]-4-yl)(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanone (7i). A pale yellow solid: mp 252-254 oC; IR (KBr) νmax 1739 (C=O), 1598 (C=CH), 1216 (C-O-C), 691 (C-F) cm-1; 1H NMR (CDCl3, 400 MHz) δ 2.38 (s, 3H, Me-H), 7.18-7.19 (m, 1H, Ar-H), 7.20-7.58 (m, 10H, Ar-H), 7.62-7.95 (m, 6H, Ar-H, H-1’’), 7.98-7.90 (m, 3H, Ar-H, H-2’’), 8.13 (d, J = 8.0 Hz, 1H, Ar-H), 8.34 (s, 1H, pyrazole-H); 13C NMR (CDCl3, 100 MHz) δ 22.7, 112.6, 119.2, 120.1, 123.2, 123.9, 124.3, 124.4, 125.0, 125.3, 125.8, 126.0, 126.0, 126.1, 126.9, 127.7, 127.8, 128.8, 128.6, 129.3, 129.5, 130.0, 130.2, 130.7, 131.3, 131.6, 134.9, 136.6, 138.4, 139.5, 146.3, 147.4, 154.8, 184.6; ESI MS m/z (rel int): 581 (M+H)+(100). Anal. Calcd for C37H25FN2O2S: C, 76.53; H, 4.34; N, 4.82. Found: C, 76.49; H, 4.31; N, 4.78.
(E)-(4-(naphthalen-1-yl)phenyl)(3-(2-(1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl)vinyl)benzofuran-2-yl)methanone (7j). A pale yellow solid: mp 268-270 oC; IR (KBr) νmax 1741 (C=O), 1600 (C=CH), 1232 (C-O-C) cm-1. 1H NMR (CDCl3, 400 MHz) δ 7.19-7.21 (m, 1H, Ar-H), 7.29-7.34 (m, 2H, Ar-H), 7.41-7.81 (m, 16H, Ar-H, H-1’’), 7.90-7.96 (m, 3H, Ar-H), 8.04 (d, J = 16.5 Hz, 1H, H-2’’), 8.16 (d, J = 8.0 Hz, 1H, Ar-H), 8.26 (d, J = 8.5 Hz, 2H, Ar-H), 8.37 (s, 1H, pyrazole-H); 13C NMR (CDCl3, 100 MHz) δ 112.7, 115.5, 119.2, 119.4, 120.2, 120.4, 123.1, 124.3, 125.0, 125.3, 125.6, 126.0, 126.1, 126.4, 126.9, 127.0, 127.4, 127.7, 128.3, 128.4, 128.8, 129.3, 129.5, 129.5, 129.9, 130.1, 130.1, 131.2, 131.3, 133.8, 139.1, 139.2, 139.5, 145.5, 154.8, 171.7, 185.5; ESI MS m/z (rel int): 599 (M+H)+(100). Anal. Calcd for C40H26N2O2S: C, 80.24; H, 4.38; N, 4.68. Found: C, 80.21; H, 4.42; N, 4.64.
Antibacterial activity
The synthesized novel compounds 5 and 7a-j were screened for their antibacterial activity against different types of bacterial strains, they are Gram negative bacterial strains of Pseudomonas aeruginosa and Escherichia coli, Gram positive bacterial strains of Bacillus subtilis and Staphylococcus aeureus at a concentration of 50 µg/mL . The cultures were diluted with 5% autoclaved saline and the final volume was made with concentration approximately 105-106 CFU/mL. The synthesized compounds were diluted in acetone for antibacterial biological assays. For agar disc diffusion method [40], the liquid form of test compound was soaked on to the disc and then allowed to air dry, such that the disc gets completely saturated with test compound. The saturated chemical discs were introduced onto the upper layer of the medium evenly loaded with the bacteria. The discs were dipped in different chemical samples, were placed over the evenly spread bacterial nutrient media and incubated at 37 oC for 24 to 48 h for better inhibition of bacteria. The zones of inhibition were measured after 24 to 48 h. All the experiments were carried out in triplicates and the results were expressed as zone of inhibition in mm. The zones of inhibition of synthesized compounds were compared with the zone of inhibition of standard antibiotic concentration (50 µg/mL). The antibacterial activity was evaluated and the results are presented in Fig. 2.
Antifungal activity
The antifungal activity of synthesized compounds was tested against three pathogenic fungi, namely Fusarium oxysporum, Aspergillus niger, and Penicillium italicum, by the poison plate technique at a concentration of 50 µg/mL. Three kinds of fungi were incubated in PDA at 25±1 °C for 5 days to get new mycelium for antifungal assay, then a mycelia as disks of approximately 0.45 cm diameter cut from the culture medium were picked up with a sterilized inoculation needle and inoculated in the center of PDA plate. Test compounds were dissolved in acetone (10 mL) then added to the Potato Dextrose Agar medium (PDA, 90 mL). The final concentration of compounds in the medium was adjusted to 50 µg/mL. The inoculated plates were incubated at 25±1 °C for 5 days. Acetone was diluted with sterilized distilled water and used as control, while clotrimazole (50 µg/mL) was used as standard control for each treatment three replicates of experiments were carried out. The radial growth of the fungal colonies was measured on the sixth day.

