











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
The endemic regions of malaria and tuberculosis - the most widespread parasitic and bacterial infections worldwide - overlap to a very considerable extent. [1-3] This is especially so in sub-Saharan Africa, where a large part of the population is exposed to both infections. However, the interactions between the two diseases is not well understood, and there is little or no clinical data on the outcomes of co-infected patients.[2,4] The effects on the host immune system of both infections is well understood, and the effects should interfere with the capability of the immune system to handle an infection by the other pathogen. This effect is probably strongest for malaria, as infection by plasmodium lowers, for example, the number of T lymphocytes, the proportion of T lymphocytes that are TH cells, and the ratio of TH to T-suppressor cells, all of which should be detrimental to the immune response to a Mycobacterium tuberculosis infection. [3] There are mouse model results that show that Plasmodium infection increases the virulence of M. tuberculosis and this may indicate that malaria could trigger a chronic M. tuberculosis infection in man to become symptomatic. [4,5] The latter has the potential to be a significant factor in tuberculosis epidemiology as many of the estimated 2 billion people with chronic tuberculosis live in malaria-endemic regions of the world.
Malaria is the disease caused by infections of a number of Plasmodium species, of which P. falciparum and P. vivax are responsible for the large majority of all deaths. The parasite is spread between human hosts by mosquitoes of the genus Anopheles, and in many endemic regions large parts of the population experience multiple infections each year, leading to partial immunity. [6] As an effect, young children who have not yet acquired immunity, women whose malaria immunity is suppressed during pregnancy, and travelers from regions without endemic malaria are most at risk. Much effort has been spent on curbing the transmission of malaria, utilizing bed nets, insect repellent spraying and eliminating sources of standing water where mosquitoes breed. However, chemotherapy of both routine and severe malaria is, and will continue to be, a vital part of the global fight against malaria. [7]
Malaria drugs fall in two general categories, quinolines and artemisinins. The quinolines include the natural product quinine, still used for severe malaria in many parts of the world, and synthetic quinolines of mainly two classes: aminoquinolines and methanolquinolines. The artemisinins are semisynthetic derivatives of the natural product artemisinin, extracted from the roots of the Chinese shrub Artemisia annua, or sweet wormwood. The most successfully deployed aminoquinoline anti-malarial drug is chloroquine, see Fig. 1. It was used massively on a global scale for decades after its introduction in the 1940s. However, the widespread use has led to increasing resistance, and now most malaria-endemic parts of the world display almost complete chloroquine resistance.[8] Resistance has developed towards all classes of anti-malarials, and some isolates are resistant against all clinically used drugs. [7,9] As a consequence of spreading resistance and in order to hamper further development of resistance, the modern gold standard for malaria chemotherapy is considered to be artemisinin combination therapies, ACTs, where an artemisinin derivative is given in combination with a quinoline drug. [10]

Tuberculosis is caused by infection of Mycobacterium tuberculosis. The disease affects the lungs most often but may spread beyond the pulmonary tract, often resulting in very severe symptoms. Around 90 % of all infected individuals never develop any symptoms and it has been estimated that a full third of the world’s population carry latent tuberculosis infections. [10] Latent infections can become active, often when the immune system is suppressed or challenged by for example starvation, other disease, medications or narcotics or alcohol use. HIV/AIDS often leads to active tuberculosis, and HIV screening is an integral part of modern tuberculosis treatments.
Modern tuberculosis treatments always rely on multi-drug combinations taken for several months. The standard regimen recommended by the WHO and implemented globally is a combination of isoniazid, rifampicin, pyrazinamide, and ethambutol (Fig. 2) for two months to bring the infection under control, and isoniazid and rifampicin for a further four months to eliminate all bacteria in the patients. [11] Despite the use of long treatment regimens and multi drug-drug cocktails, drug resistant tuberculosis is endemic in many countries, and the treatment of extensively drug-resistant tuberculosis with third and fourth line drugs is complicated and costly, and has high rates of failure. [10]
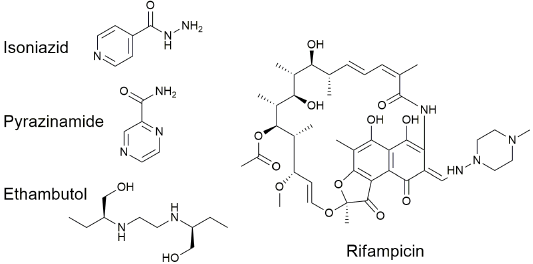
Fig. 2 Structures of the anti-mycobacterial drugs isoniazid, pyrazinamide, ethambutol, and rifampicin, which are used as first line treatment of active tuberculosis. [11]
Considering the problems with drug resistance against both malaria and tuberculosis drugs, it is no surprise that much effort has been spent on drug discovery as well as structural modifications of existing drugs to overcome drug resistance. For both diseases, organometallic compounds have been evaluated as potential drugs. The ferrocene-containing chloroquine analogue ferroquine (Fig. 3) exhibits excellent anti-plasmodial activity and no cross resistance with chloroquine. [12-14] Ferroquine has undergone stage IIB clinical trials and is set to enter further clinical trials. Other organometallic compounds that have been evaluated for anti-plasmodial activity include organometallic moieties coordinated to chloroquine and aminoquinoline-based ligands coordinated to a variety of organometallic moieties. [15-27] There is considerably less literature on organometallic anti-mycobacterial agents, but compounds that have been investigated for such activity include cyrhetrenyl and ferrocenyl thiosemicarbazones, phenylated palladium complexes, as well as ruthenium and rhodium half sandwich complexes of silylated aminoquinolines, and various aryl-tin complexes. [28-32]
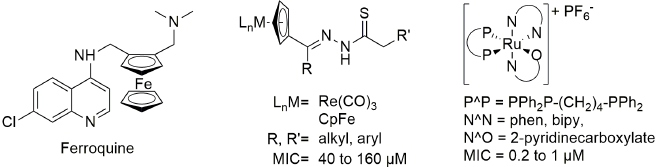
Fig. 3 Structure of the organometallic anti-malarial drug candidate ferroquine, and examples organometallic and coordination complexes evaluated for anti-mycobacterial activity. [12, 21, 26]
In this study, we have set out to investigate the possibilities of aminoquinoline hybrids with pharmacophores from tuberculosis drugs to expand our family of aminoquinoline-based ligands for half sandwich complexes that may function as potential anti-plasmodial and anti-mycobacterial agents. Pyrazinamide offers an intriguing choice for this purpose, as the pyrazine and the amide nitrogens offer a chelating coordination moiety with a similar bite as previous N^N-coordinating amino-pyridyl and amino-imidazole ligands investigated in our laboratory.
Results and discussion
Synthesis, characterization and molecular structures. Reaction of N1-(7-chloroquinolin-4-yl)ethylene diamine with one equivalent of pyrazine carbonyl chloride, prepared from pyrazine carboxylic acid and thionyl chloride, in dichloromethane in the presence of triethylamine gave N-(2-((7-chloroquinolin-4-yl)amino)ethyl)pyrazine-2-carboxamide, L, in good yield (Scheme 1). The pyrazine carbonyl chloride was found to decompose even when stored under dry nitrogen in a refrigerator; the originally white crystals turned first pink and then purple over a period of a few days, and the 1HNMR spectrum showed approximately 20 % of unidentified decomposition products relative to the intact pyrazine carbonyl chloride. However, pyrazine carbonyl chloride that was pure to the limits of detection by 1HNMR could be retrieved by vacuum sublimation of the pink or purple material, and was used immediately in further reactions.
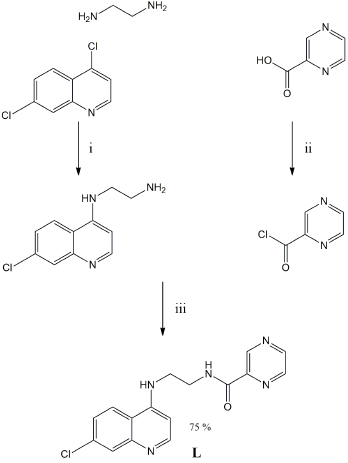
Scheme 1 Synthesis of L. Reagents and conditions: i) 130 °C, 8 h, neat. ii) SOCl2, reflux, followed by vacuum sublimation. iii) CH2Cl2, Et3N, 0 °C 3 h, RT overnight.
Scheme 2 Synthesis of L. Reagents and conditions: i) 130 °C, 8 h, neat. ii) SOCl2, reflux, followed by vacuum sublimation. iii) CH2Cl2, Et3N, 0 °C 3 h, RT overnight.
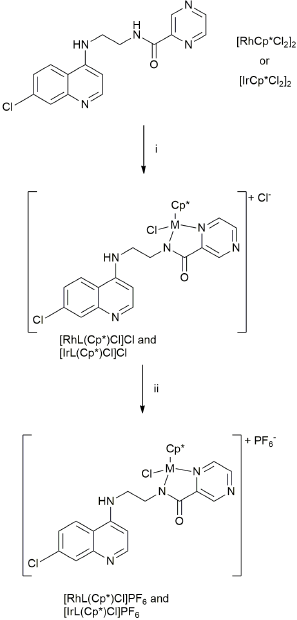
Reaction of L with 0.5 equivalents of [RhCp*Cl2]2 or [IrCp*Cl2]2 in a mixture of dichloromethane and methanol gave the complexes [Rh(L)(Cp*)Cl]Cl and [Ir(L)(Cp*)Cl]Cl. For both complexes, the Cl-anion was exchanged for PF6- by treating solutions of the complexes with AgPF6, without prior isolation of the chloride salts (precursor complexes). Single crystals suitable for X-ray diffraction were obtained by slow evaporation of dichloromethane/hexane solutions of the hexafluorophosphate salt of [Rh(L)(Cp*)Cl]+ and of the chloride salt of [Ir(L)(Cp*)Cl]+.
Reaction between L and [Ru(p-cymene)Cl2]2 under various conditions, including activation by silver salts either before the addition of L or in the presence of the ligand, gave complicated mixtures of products that could not be purified by column chromatography or recrystallization, and which tended to decompose into even more complicated mixtures both as a solid and in solution in organic solvents.
The molecular structures of the cations of [Rh(L)(Cp*)Cl]PF6 and [Ir(L)(Cp*)Cl]Cl are shown in Fig. 4; selected bond lengths and angles are collated in Table 1, and crystallographic details are listed in Table 2. The two structures are closely related and exhibit similar bond distances and angles between the coordinated ligands, for example M-Npyrazole, M-Namido and M-Cl distances are 2.082(5), 2.104(6), and 2.444(2) Å for [Rh(L)(Cp*)Cl]PF6 while the same distances are 2.110(6), 2.090(6), and 2.404(2) Å in [Ir(L)(Cp*)Cl]Cl. However, the conformations of the non-coordinated parts of L differ significantly between the two structures. In the iridium complex, the quinoline and pyrazine heterocycles are essentially coplanar, with the dihedral angle between the two ring systems being 4°, while in the rhodium complex the quinoline is folded back by almost 180° relative to the iridium structure, and the same quinoline and pyrazine planes make a dihedral angle of 48°. In the crystal packing of the rhodium complex, the quinolines on adjacent molecules are stacked in an antiparallel fashion, while in the packing of the iridium complex the quinoline is stacked parallel with a pentamethylcyclopentadienyl ring of a neighboring molecule and the pyrazine rings of adjacent molecules are stacked slightly offset with respect to each other. The structures of both complexes closely resemble published complexes with pyridyl-amides coordinated in the same fashion. To the best of our knowledge, the present structures are the first reported structures of a pyrazine-amide coordinated to a rhodium or iridium atom.
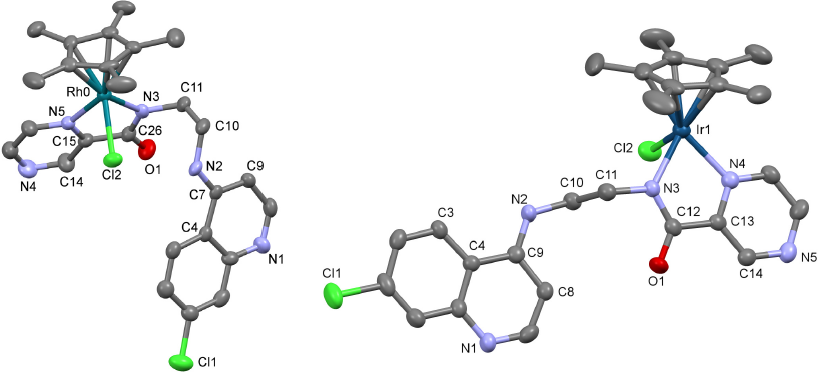
Fig. 4 Fig. 4. Structures of the cations of [Rh(L)(Cp*)Cl]PF6 (left) and [Ir(L)(Cp*)Cl]Cl (right), showing the atom numbering schemes. Thermal ellipsoids are plotted at the 30% probability level. Hydrogen atoms have been omitted for clarity.
Table 1 Selected bond lengths and angles for [RhL(Cp*)Cl]PF6 and [IrL(Cp*)Cl]Cl
aThe pyrazine plane is defined by the pyrazine carbons and nitrogens, as implemented in Mercury. The quinoline plane is similarly defined as the quinoline carbons and nitrogen, while the Cp* plane is defined as the carbons in the ring, i.e. excluding the methyl carbons
Table 2 Selected crystal data for [RhL(Cp*)Cl]PF6 and [IrL(Cp*)Cl]Cl
aR1 = Σ||Fo| - |Fc|/Σ|Fo|
bwR2 = [Σw(Fo2 - Fc2)2/Σw(Fo2)2]1/2, where w = 1/[σ2(Fo2) + (aP)2 + bP] where P = (Fo2 + 2Fc2)/3.
Biological results. Ligand L and the complexes [Rh(L)(Cp*)Cl]PF6 and [Ir(L)(Cp*)Cl]PF6 were evaluated in vitro for antimicrobial activity against the NF54 strain of Plasmodium falciparum and against the H37Rv strain of Mycobacterium tuberculosis. The results of the in vitro studies are presented in Table 3. Ligand L and complex [Rh(L)(Cp*)Cl]PF6 were active against P. falciparum while [Ir(L)(Cp*)Cl]PF6 did not exhibit any anti-plasmodial activity at the highest concentration tested. However, the activities of L and [Rh(L)(Cp*)Cl]PF6 were moderate - the IC50 values are approximately 70 and 50 times higher than for the reference drug chloroquine, respectively. Smith et al. have shown that the organometallic starting materials, [RhCp*Cl2]2 and [IrCp*Cl2]2, are toxic to the strain of Plasmodium falciparum used in this evaluation, but only at concentrations significantly higher than those employed in this study.[20] It is therefore reasonable to assume that the displayed anti-plasmodial activity is related to the amino-quinoline pharmacophore in L rather than to some metal-specific mechanism.
Table 3 Anti-plasmodial and anti-mycobacterial activity for L, [RhL(Cp*)Cl]PF6 and [IrL(Cp*)Cl]PF6. The antiplasmodial activity is expressed as IC50 values against the NF54 strain of P. falciparum, the anti-mycobacterial activity as the minimum inhibitory concentration, MIC99, against the H37Rv strain of Mycobacterium tuberculosis.
aIC50, the concentration that inhibits parasite growth by 50 %.
bMIC99, the lowest concentration that inhibits mycobacterial growth by 99 %.
cNo activity detected at the highest concentration tested.
All three compounds did inhibit the growth of M. tuberculosis, but only at high concentrations. Ligand L is almost a thousand times less active than the reference drug isoniazid and the activities of the complexes [Rh(L)(Cp*)Cl]PF6 and [Ir(L)(Cp*)Cl]PF6 are approximately 50 times less active. It is noteworthy that the organometallic complexes are 16 times more active than the free ligand, pointing to a potential benefit of including half-sandwich moieties in anti-mycobacterial drugs.
Summary and Conclusions
The aminoquinoline - pyrazinamide hybrid N-(2-((7-chloroquinolin-4-yl)amino)ethyl)pyrazine-2-carboxamide and its complexes with rhodium and iridium pentamethylcyclopentadienyl moieties have been synthesized and characterized. The molecular structures of the hexaflourophosphate salt of the rhodium complex and the chloride salt of the iridium complex have been studied by X-ray diffraction, and the two complexes show very similar coordination geometries. The ligand and the rhodium complex exhibit moderate anti-plasmodial activity, and the ligand and both complexes exhibit anti-mycobacterial activity, albeit at high concentration. It should be noted that the organometallic complexes are more than one order of magnitude more active against M. tuberculosis than the free ligand in in vitro experiments, which warrants further studies.
Experimental
General. All chemicals were purchased from Sigma Aldrich, except [Rh(Cp*)Cl2]2 and [Ir(Cp*)Cl2]2 which were obtained from Strem. All chemicals were used as received. N-(7-chloroquinolin-4-yl)ethane-1,2-diamine [31] and pyrazine-2-car-bonyl chloride [32] were synthesized according to literature procedures. 1H NMR spectra were recorded on a Varian Inova 500 MHz spectrometer at ambient temperature using the residual solvent peak as internal standard. High resolution mass spectra (HRMS) were obtained on an LTQ Velos Orbitrap mass spectrometer in ESI positive mode and a resolution (Rs) of 30 000; raffinose and sodium was added for lockmass correction.
Syntheses. N-(2-((7-chloroquinolin-4-yl)amino)ethyl)pyrazine-2-carboxamide, L. N-(7-chloroquinolin-4-yl)ethane-1, 2-diamine (559 mg, 2.53 mmol) was suspended in a dichloromethane (200 ml) solution of triethylamine (0.529 ml, 3.8 mmol) and the suspension was added to an ice cold solution of freshly sublimated pyrazine-2-carbonyl chloride (360 mg, 2.53 mmol) in dichloromethane (20 ml). The solution was stirred overnight, during which it was allowed to reach room temperature. An off white precipitate was filtered off and washed with hexanes to yield 574 mg of L. A second crop could be recovered by addition of petroleum ether to the filtrate, filtering off the yellow precipitate formed and redissolving it in dichloromethane. The solution was washed with saturated sodium bicarbonate solution, died and filtered before it was concentrated to yield a further 50 mg of L. Total yield: 624 mg, 1.90 mmol, 75 %. 1H-NMR (d6-DMSO, 500 MHz) 9.24 (t, 1H, J=6.0Hz, pyrazole-H), 9.21 (d, 1H, J=1.3Hz, CCl-CH-C), 8.88 (d, 1H, J=2.4Hz, pyrazole-H), 8.74 (dd, 1H, J=1.5Hz, J=2.3Hz, CCl-CH-CH), 8.41 (d, 1H, J=5.4Hz, Nquinoline-CH-CH), 8.22 (d, 1H, J=9.0Hz, pyrazole-H), 7.79 (d, 1H, J=2.1Hz, CCl-CH-CH), 7.47 (m, 2H, quinolinyl-NH-CH2 and CO-NH), 6.67 (d, 1H, J=5.4Hz, Nquinoline-CH-CH), 3.62 (q, 2H, J=6.4Hz, quinolinyl-NH-CH2), 3.48 (dd, 2H, J=6.2Hz, J=12.2Hz, CO-NH-CH2) HRMS (m/z amu) 328.09677 (M+H+) (obsd.) 328.09596 (calc.)
(N-(2-((7-chloroquinolin-4-yl)amino)ethyl)pyrazine-2-carboxamide)(cyclopentadienyl) chloro-rhodium hexafluorophosphate, [Rh(L)(Cp*)Cl]PF6 . Ligand L (45.8 mg, 0.14 mmol) and [Rh(Cp*)Cl2]2 (43.5 mg, 0.070 mmol) were dissolved in CH2Cl2/MeOH (1:1, 20 ml). After stirring for 1 h at room temperature, AgPF6 (1.37 ml of a 102 mM solution in methanol) was added, and after stirring a further 30 minutes the solution was filtered through a plug of Celite; the Celite was washed with CH2Cl2/MeOH (1:1, 10 ml) and the volume of the filtrate was reduced in vacuo to ~ 5 ml. The product, [Rh(L)(Cp*)Cl]PF6, was isolated as a red powder by precipitation with ether. Yield 40 mg, 0.054 mmol, 38 %. 1H-NMR (MeOH-d4:CDCl3, 2:1, 500 MHz) 9.12 (s, 1Hz, CCl-CH-C), 8.93 (d, 1H, J=2.1Hz, pyrazole-H), 8.78 (d, 1H, J=2.3Hz, CCl-CH-CH), 8.20 (d, 1H, J=7.0Hz, pyrazole-H), 8.12 (d, 1H, J=8.7, Hz pyrazole-H), 7.73 (s, 1H, CCl-CH-CH), 7.50 (d, 1H, J=9.1Hz, Nquinoline-CH-CH), 6.83 (d, 1H, J=7.2Hz, Nquinoline-CH-CH), 4.16 (m, 1H, CO-NH-CHH), 3.86 (m, 2H, quinolinyl-NH-CH2), 3.56 (m, 1H, CO-NH-CHH), 1.74 (s, 15H, Cp*). HRMS (m/z amu) 600.08098 (M +) (obsd.) 600.07987 (calc.)
(N-(2-((7-chloroquinolin-4-yl)amino)ethyl)pyrazine-2-carboxamide)(cyclopentadienyl) chloro-iridium hexafluorophosphate, [IrL(Cp*)Cl]PF6 . This complex was synthesized from L (45.7 mg, 0.139 mmol) and [Ir(Cp*)Cl2]2 (55.5 mg, 0.0697 mmol) by the same procedure as [Rh(L)(Cp*)Cl]PF6. Yield: 36 mg, 0.044 mmol, 31 %. 1H-NMR (MeOH-d4:CDCl3, 2:1, 500 MHz) 9.11 (s, 1H, CCl-CH-C) 8.86 (s, 1H, pyrazole-H) 8.75 (dd, 1H, J=1.1Hz, J=3.2Hz, CCl-CH-CH) 8.25 (d, 1H, J=7.1Hz, pyrazole-H) 8.10 (d, 1H, J=9.1Hz, pyrazole-H) 7.74 (d, 1H, J=1.9Hz, CCl-CH-CH) 7.54 (dd, 1H, J=2.0Hz, J=9.0Hz, Nquinoline-CH-CH) 6.88 (d, 1H, J=7.2Hz, Nquinoline-CH-CH) 3.87 (dd, 2H, J=4.4Hz, J=6.8Hz, quinolinyl-NH-CH2) 3.50 (td, 1H, J=3.9Hz, J=8.5Hz, CO-NH-CHH) 3.43 (m, 1H, CO-NH-CHH) 1.74 (s, 15H, Cp*). HRMS (m/z amu) 688.1368 (M +) (obsd.) 328.1350 (calc.)
Crystallography. The X-ray intensity data for [Rh(L)(Cp*)Cl]PF6 and [Ir(L)(Cp*)Cl]Cl were measured on a Bruker Apex II CCD and an Oxford Diffraction CCD diffractometer, respectively, at room temperature using Mo Kα radiation (λ=0.71073 Å). The software SMART [33] was used for collecting frames of data, indexing reflections and determination of lattice parameters. The collected frames were then processed for integration by the SAINT program, [33] and an empirical absorption correction was applied using SADABS. [34] The structures were solved by direct methods (SIR 97) [35] and subsequent Fourier syntheses and refined by full-matrix least squares on F2 (SHELXL-2014) [36] using anisotropic thermal parameters for all non-hydrogen atoms. The aromatic, methylene and methyl H atoms were placed in calculated positions and refined with isotropic thermal parameters U(H) = 1.2 Ueq(C) or U(H) = 1.5 Ueq(C-Me), respectively, and allowed to ride on their carrier carbons whereas the imine H atom in [Ir(L)(Cp*)Cl]Cl was located in the Fourier map and refined isotropically [U(H) = 1.2 Ueq(N)]. The crystal of [Rh(L)(Cp*)Cl]PF6 was found to be twinned, and was refined as a three-component twin. Selected crystal data and details of the data collections for the two compounds are reported in Table 2. Crystallographic data for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre, CCDC, 12 Union Road, Cambridge CB21EZ, UK. Copies of the data can be obtained free of charge on quoting the depository number CCDC-1538595 ([RhL(Cp*)Cl]PF6), 1538596 ([IrL(Cp*)Cl]Cl) (Fax:+44-1223-336-033; E-Mail: deposit@ccdc.cam.ac.uk, http://www.ccdc.cam.ac.uk)
Determination of in vitro anti-plasmodial activity. Ligand L and complexes [Rh(L)(Cp*)Cl]PF6 and [Ir(L)(Cp*)Cl]PF6 were evaluated against the chloroquine sensitive strain NF54 of P. falciparum. Continuous in vitro cultures of asexual erythrocyte stages of P. falciparum were maintained using a modified method of Trager and Jensen.[37] Quantitative assessment of antiplasmodial activity in vitro was determined via the parasite lactate dehydrogenase assay using a modified method described by Makler.[38] The samples were tested in triplicate on one occasion. The test samples were prepared as a 20 mg/ml stock solution in 100% DMSO. Stock solutions were stored at -20 ºC. Further dilutions were prepared on the day of the experiment. Chloroquine diphosphate (CQ) was used as the reference drug in all experiments. A full dose-response was performed for all compounds to determine the concentration inhibiting 50% of parasite growth (IC50-value). Samples were tested at a starting concentration of 10 μg/ml, which was then serially diluted two-fold in complete medium to give 10 concentrations with the lowest concentration being 0.02 μg/ml. Chloroquine was tested at a starting concentration of 100 ng/ml. The highest concentration of solvent to which the parasites were exposed had no measurable effect on the parasite viability (data not shown). The IC50-values were obtained using a non-linear dose-response curve fitting analysis via Graph Pad Prism v.4.0 software.
Determination of minimal inhibitory concentration against Mycobacterium tuberculosis. Ligand L and complexes [Rh(L)(Cp*)Cl]PF6 and [Ir(L)(Cp*)Cl]PF6 were evaluated against the H37Rv strain of Mycobacterium tuberculosis. MICs were determined by the broth microdilution method using 2-fold dilutions of the compounds in 96-well microtitre plates. [39] Briefly, M. tuberculosis H37Rv cells were grown in Middlebrook 7H9 media supplemented with 0.2 % glycerol, Middlebook OADC enrichment and 0.05 % Tween 80 (7H9) to OD600nm of 0.2-0.3 (~106 CFU/ ml) and diluted 1:500 in 7H9 media to be used as inoculum. Compounds were dissolved in DMSO and were diluted to a concentration of 640 μM in 7H9 for testing. Fifty μl of 7H9 media was dispensed in a 96 well round-bottom plate in each well except in the top row in which 100 μl of media was added to the first well (control), the appropriate concentration of the DMSO diluted in 7H9 to the second well (control) and the respective compounds (640 μM) in duplicate in the next six wells. Two-fold dilutions were carried out using a multichannel pipette from the top row to the last row from which 50 μl was discarded. All the wells in the plate were inoculated with 50 μl of the diluted H37Rv culture. The plates were incubated (37 °C, 95% humidity) and read visually using an inverted mirror after 7 and 14 days. The standard drugs, rifampicin, isoniazid and streptomycin were used as positive controls. The MIC was determined as the lowest concentration that prevented growth of the bacterial cells.

