



![Synthesis and Structural Characterization of [BmMeBenz]2Ca(THF)2: Ca•••H-B interactions in a Sulfur-Rich Coordination Environment](/img/es/prev.gif)







 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Aromaticity is arguably one of the most important and fascinating concepts in organic chemistry [1-2]. The understanding of this key concept has provided the most fruitful interplay between experiment and theory in chemistry [3]. In organic chemistry textbooks, aromaticity is associated with cyclic planar conjugate molecules in which the number of π electrons is 4n+2 (the Hückel’s rule). The archetypal example of an aromatic molecule is benzene [3]. Using the Huckel’s rule, aromaticity can be assigned to any conjugated cyclic molecule formed exclusively by main group atoms with a formal sp2 hybridization since the numbers of π electrons in these atoms can be easily obtained; however, when the cycle contains a transition metal fragment the application of Huckel’s rule is not so straightforward. In order to circumvent this difficulty, the aromaticity in metallacycles is usually related to compounds in which an organic fragment of an aromatic molecule is replaced by an isolobal organometallic analogue. In this way we can make a bridge between the classical organic definition of aromaticity (the Huckel’s rule) and their occurrence in organometallic chemistry. The appealing of this equivalence between organic and inorganic chemistry is the fact that they rest on a simple electron count problem that can be applied without the use of more elaborate quantum chemical calculations.
In a premonitory article, in 1979, Thorn and Hoffmann [4] theoretically predicted the existence of metallabenzenes. This prediction rapidly increased the interest on this class of metallacycles, and the first osmabenzene was synthesized in 1982 [5]. From that moment, the chemistry of aromatic me-tallabenzenes shows a major expansion in terms of new metallabenzenes, new synthetic methods, and more metal centers [6-9]. Up to date, over 30 varieties of metallabenzenes have been synthesized and characterized [10]. The largest families of metallabenzenes are the osmabenzenes (1) and iridabenzenes (2) [6-8], whose stability was associated to their apparent aromaticity.
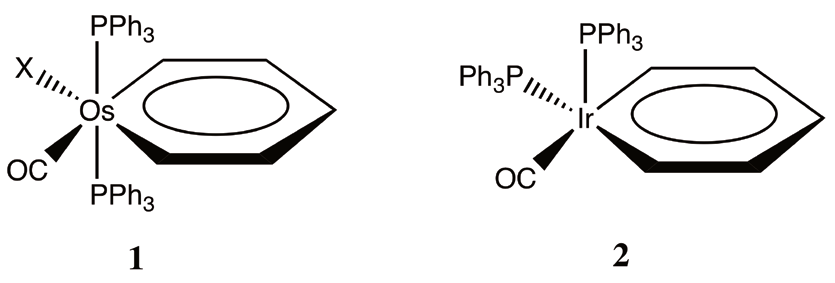
Metallabenzenes are classified as aromatic molecules on the basis of the observed properties: they are planar (or quasi-planar) cyclic structures that exhibit bond-length equali-zation, show aromatic deshielding in 1H NMR spectroscopy and, recently, it was demonstrated that osmabenzene follows electrophilic aromatic substitution reactions such as nitration, chlorination and bromination [10]. Nevertheless, some unusual reactions for aromatic molecules like cycloaddition and rearrangements to form cyclopentadienyl complexes are observed. Moreover, the anisotropy of the transition metal center affects the 1H NMR spectra, especially for hydrogen at ortho position in which unusual proton chemical shift between 12-25 ppm are observed. These latter observations suggest some important differences between organic aromatic molecules and their transition metal analogues.
An important question whether is it possible or not to quantify the aromaticity? Aromaticity is a concept that results fundamental for the understanding of the physical and chemical properties of conjugated molecules, related to the manifestation of electron delocalization in a closed circuit that resulting in a quite substantial energy stabilization, bond length equalization, unusual reactivity and particular magnetic properties. However, the fact that the degree of aromaticity is not directly measurable, introduces a source of considerable confusion and arbitrariness to going beyond the qualitative Huckel’s rule. Indeed, throughout the literature there is not a unique definition underlying the quantification of aromaticity. Among the most popular ways proposed for quantification of aromaticity we can mention: (i) the Harmonic Oscillator Model of Aromaticity (HOMA) index developed by Kruszewski and Krygowski [11-12], which is a geometric based descriptor that looks to capture the bond length equalization effect of aromatic cycles, (ii) the Aromatic Stabilization Energy (ASE) [13-14], and (iii) the Nucleus Independent Chemical Shift (NICS) [15-17] which is defined as the negative value of the isotropic absolute shielding computed at the ring center, NICS(0), or 1 Å from the plane of the NICS(0) point, NICS(1). The previous list, which is very far from being exhaustive, reveals that the definition of aromaticity is an open problem that can be approximated from different non-coincident perspectives. For a more complete list, we invite the readers to consult some reviews on this field (see for instance the special issue of Chemical Review on Aromaticity in 2005 [18] and the special issue of Phys. Chem. Chem. Phys. celebrating the 150 years of the Kekule structure of benzene [19]). Before continuing, it is important to remark that, like many other concepts in chemistry, the definition of aromaticity cannot be right or wrong in an absolute way; it can only be useful or not, and in some sense, it is a matter of individual preference at the moment of choosing a particular definition for a given problem. Moreover, the notion of aromaticity is necessary in chemical education and theoretical chemistry, because it provides and exceptional theoretical framework, in terms of which we can rationalize patterns that are observed in the experiments.
Theoretical chemists have tried to explain the stability and reactivity of metallacycles focusing their attention on the degree of aromaticity by calculating theoretical descriptors of aromaticity [20-27]. The first theoretical study advocated to quantify the aromaticity of metallacycles was published in 1993 by Chamizo, Morgado and Sosa [20]. From the absolute hardness, calculated by employing the Extended Hückel (EHT) method, the authors concluded that iridabenzenes (2) are much less aromatic than benzene [20]. In 2004, De Proft and Geerling [21] performed aromatic stabilization energy calculations [13-14] to study the degree of aromaticity of a platinabenzene. They found that platinabenzene was slightly less aromatic than benzene [21]. Iron et. al. [22] study the aromaticity of 10 metallabenzenes using the Nucleus Independent Chemical Shift (NICS) [15-17], concluding that their aromaticity is lower than benzene. In the latter study, it was clear that the NICS values depend strongly on the nature of both the metal atom and their ligands [22]. In 2007, Fernandez and Frenking [23] used an energy decomposition analysis method [24] to analyze the aromatic character of 21 metallabenzenes. In the later work, 5d metallabenzenes shows larger aromaticity than the 4d analogs. In 2008, Periyasami et. al. [25] used the same set of metallabenzenes of Fernandez and Frenkling [23] to study their aromatic character using magnetic descriptors. This work shows that Ir and Rh 18-electron complexes should be considered aromatics; on the contrary, Os and Ru 16-electron systems should not. In all cases, it has been found that aromaticity of metallabenzenes is reduced in comparison with benzene [25]. In 2012, Feixas et. al. [26] published a critical review on the theoretical works on metalloaromaticity with emphasis in all-metal aromaticity; from this latter review it is clear the lack of reliable measurements of aromaticity. More recently, in 2015, Fernandez, Frenking and Merino publish a critical evaluation of the performance of several aromatic descriptors when applied to metallabenzenes and related heterocycles.
What is clear from a critical analysis of the experimental and theoretical studies on metallabenzenes mentioned before is the fact that these compounds display many contradictory behaviors when compared with their organic analogs, and their aromaticity can be severely questionable. In order to explain these differences, some authors argue that two filled metal d orbitals are strongly involved in the delocalization and thus metallabenzenes possess eight π-electrons, four of these π electrons have Hückel aromaticity type character while the remaining two have δ symmetry and can be understood as a Mobius aromaticity. However, what is missing at this moment in the literature is a simple explanation to this aromaticity reduction in matallacycles. To shed some light on this issue we propose that metallabenzenes can be considered as structures intermediate between two extreme situations: the benzene (3) and the benzenonium ion (4).

We reach this conclusion after a careful application of isolobal analogies to the case of metallabenzenes. Even with the highly speculative character of the present proposal, we present some evidence that using this characterization of metallabenzene can explain some unusual issues of the reactivity and magnetic properties of osmabenzenes (1) and iridabenzenes (2). At the end of this work, we envision further theoretical (and even experimental) works to corroborate our hypothesis.
Aromaticity in metallabenzenes from isolobal analogies
The basic tool to establish a link between the qualitative Huckel’s rule and the aromaticity of metallacycles are the so-called isolobal analogies. Roald Hoffmann summarized the isolobal analogies in his 1981 Nobel lecture, about the relationships between the bonding and reactivity in transition metal complexes and simple organic molecules [28-29]. An excellent introduction to isolobal analogies can be found in the book of Albright, Burdett and Whangbo entitled “Orbital Interactions in Chemistry” [30]. According to Hoffmann: “Two fragments are isolobal if the number, symmetry properties, approximate energy and shape of the frontier orbitals and the number of electrons in them are similar-not identical but similar” [29]. This persuasive idea had some previous elaborations [31-35]; however, it was the Hoffmann’s group who fully exploits the isolobal analogies and finally popularizes them in organometallic chemistry [30]. The idea of isolobal analogies has proved to be a very useful concept for unifying large areas of inorganic and organic chemistry and highlighting important similarities between main group and transition metal chemistry. In this section, after a brief introduction to the isolobal analogies, we derived from it the basic rule of aromaticity in metallabenzenes.
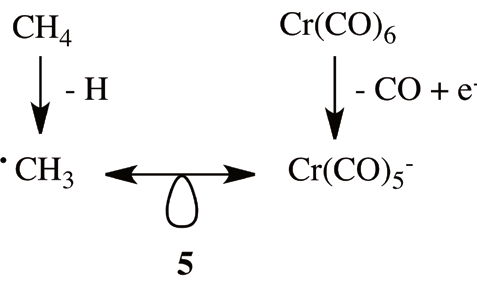
The basic idea behind the isolobal analogies is the following: an homolytic removal of a hydrogen atom from methane to give the methyl radical produces a half-filled frontier orbital of the same symmetry, and also similar energy, to one of the Cr(CO) 5 ˉ radical.
This appealing idea is summarized in 5. As is shown in 5, it is conceivable that the radical was created starting from the 18-electron complex Cr(CO)6 by removal of a CO and addition of an extra electron. Since the frontier orbitals of both radicals are similar, their chemistry also is similar. The use of this approach allows one to move vis-a-vis from a transition metal complex with a fragment, or more general a d7-ML5 fragment, to a methyl fragment in an organic molecule. A generalization of the previous rule allows one to move from an organic aromatic molecule to a related metallacycle. The key issue in 5 is the connection between the 8-electron main group specie, CH4, and the 18-electron octahedral complex, Cr(CO)6. Before continuing, it is important to keep in mind that isolobal analogies must be used exclusively in a qualitative sense.
Hereafter, we consider the application of isolobal analogies to aromatic metallabenzenes. Metallabenzenes can be considered as a transition metal fragment, MLn, interacting with the bidentade-monoanionic ligand, C5R5 -, that result from the remotion of a cationic carbine fragment to the benzene [4,6]. For electron count purposes, the C5R5 - unit is a four-electron σ donor with 4π electrons. Thus, for a metallacycle to be isolobal with benzene, a 2π electron cationic carbyne singlet fragment, CH+, must be replace by a metal analogue. Now, if as in 5 we start from a 18-electron octahedral complex, d6-ML6 (for example a Cr(CO)6 complex), and we remove three of their ligands and insert three extra electrons, we reach the conclusion that the resulting 15-electron three coordinate fragment, d9-ML3, is isolobal to the carbyne organic fragment that results from the homolytic dissociation of three hydrogens from methane. This equivalence is illustrated in 6 and results of a subsequent application of 5. Thus, a CH+ is isolobal with a d8-ML3 fragment (7). Following this line of thought, metallabenzenes are aromatic cycles in which one CH of the benzene was replaced by a d8-ML3 metallic fragment. A representative example of this kind of metallabenzenes is the family of iridabenzenes (2), ruthenabenzenes [36] and platinabenzenes [10]. However, the previous rule can’t explain the aromaticity of osmabenzenes (1).
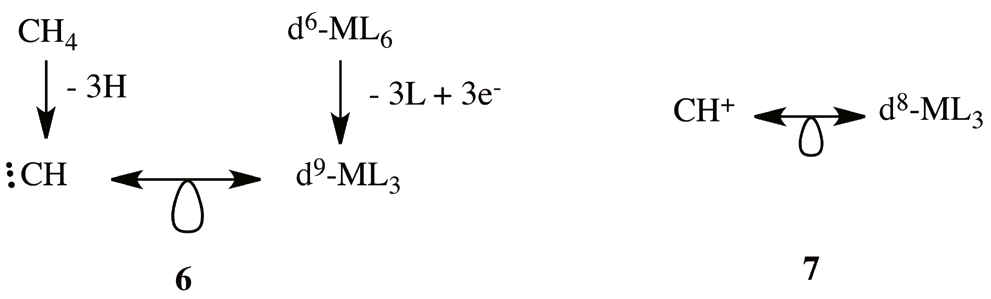
To further complicate the situation further, there is a strong caveat in the previous naïve application of the isolobal analogies [30]. For instance, if instead of the octahedral d6-ML6 complex used as starting complex to obtain 5 and 6, we use as starting point the trigonal bipyramidal complex, d8-ML5, and we remove two ligands and add two electrons, we reach the conclusion that the fragment d10-ML3 is isolobal with the carbene, CH2 (8). Removing two electrons from 8 means that a fragment d8-ML3 is isolobal with CH2 2+ (9).
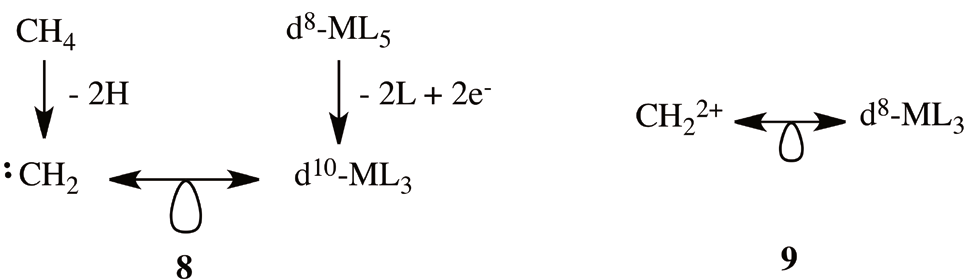
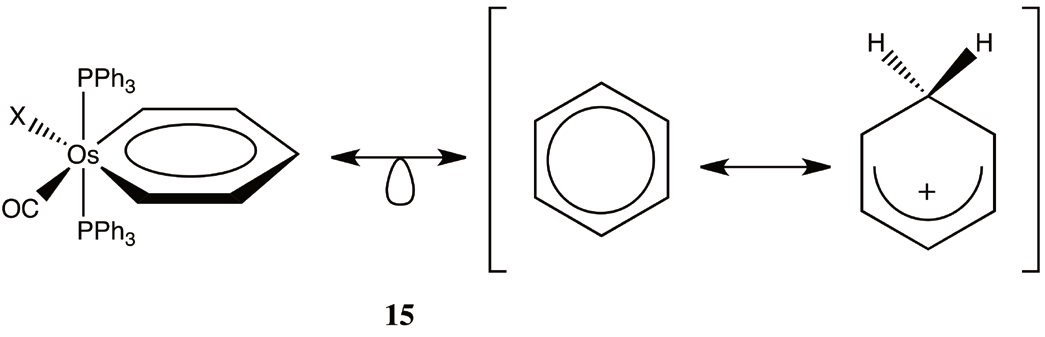
Now, we have a paradox: is d8-ML3 fragment isolobal to CH+ (7) or CH2 2+ (9). This question has a profound consequence, because if 7 is true, the iridabenzenes are isolobal to benzene, and are aromatic, but in the case of 9 is true, the iridabenzenes are isolobal to the benzenium cation (4), which is not longer aromatic. How do we discern between 7 and 9? Since equivalences 7 and 9 are derived from 6 and 8, we are going to look into more detail at the starting complex of each case. Because the starting complex of the CH+ fragment is an octahedral d6-ML6 complex (6) and for CH2 2+ is a trigonal bipyramidal d8- ML5 (7), it is important that in addition to the oxidation state of the metal and the number of ligands we look at bond angles between ligands. For a CH+ isolobal fragment we expect, based on the d6-ML6 octahedral starting complex, an angle of 90? between ligands, on the contrary, for a CH2 2+ this angle increase to 120? in the equatorial plane. The X-ray structure of the iridabenzene (2) with triphenylphosphines shows two P-Ir-P bond angles, the bond angle between the phosphines in the plane of the metallacycle, P1- Ir-P1, is 94.43?, and the bond angle between the phosphines in the plane and axial phosphines is 99.05? [37]. Larger angles are found when bulky phosphines are employed [10]. These angles indicate that the bonding pattern in these metallacycles can be conceptualized as intermediate between two extreme situations: on the one hand the benzene (3) and at the other extreme, the benzenium cation (4). We illustrate this tension in (10) using the resonance symbol (? )to illustrate a situation that result from a hybrid description between two extremes extructures. In the following, we hypothesize that this dichotomy is present in all conceivable metallabenzenes; moreover, is in some way inherent to their electronic nature.

In order to generalize our hypothesis (10), consider for instance the case of osmabenzenes (1). Osmabenzenes, are a family of Os(II) tetracoordinated fragments linked to the C5H5- organic framework. As in the case of iridabenzenes (2), if we start with two different kinds of 18-electron complexes, one arrives to a different isolobal analogy. Starting from the 18-electron octahedral complex d6-ML6 (think for instance in the Cr(CO)6 complex) and we remove two ligands and add two electrons, we reach the conclusion that a d8-ML4 complex (derived from an octahedral) is isolobal with the carbene CH2 fragment (11). From the previous analogy, the fragment osmabenzene fragment d6-OsL4 is isolobal with CH2 2+ (12). Thus, as in the case of iridabenzenes, the osmabenzenes are isolobal in this way to the benzenium cation (4).
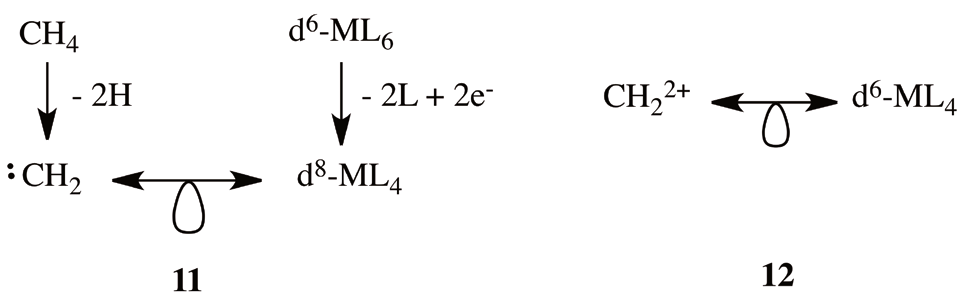
In contrast to (11), we can start from a seven-coordinate 18-electron d4-ML7 complex. Even if very uncommon, these kind of complexes could be a pentagonal bipyramid (D5h), a capped octahedral (C3v) or a capped trigonal prism (C2v) [38]. Remotion of three ligands and insertion of three extra electrons resulting in a 15-electron fragment, d7-ML4, which is isolobal to the carbyne organic fragment (13). From the remotion of an extra electron we conclude that the fragment OsL4 is isolobal with CH+ (14).
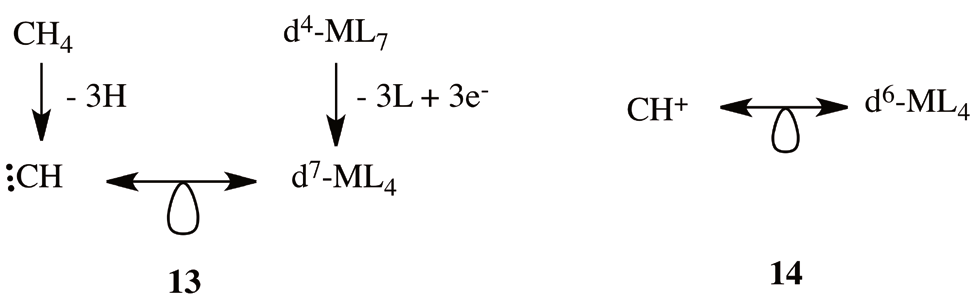
From the inspection of (12) and (14) we conclude that as in 10 osmabenzenes could be represented as in 15.
Final Remarks
From the previous section, after a detailed analysis of the isolobal analogies in iridabenzenes (10) and osmabenzenes (15), we can conclude that these structures can be represented as a combination of two extreme situations: the benzene and the benzonium ion. This conclusion is fully illustrated in 16, and is the main hypothesis of this work.
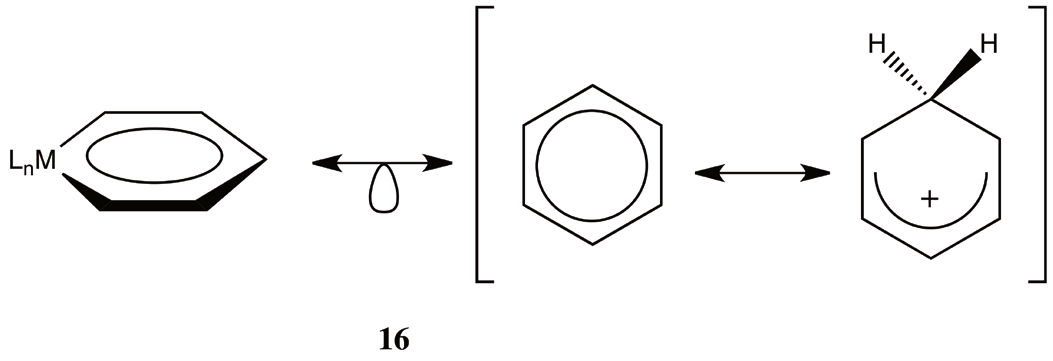
From 16 is clear that we expect for metallabenzenes, a planar ring with bond length equality, as is observed [6-8]. The fact that metallabenzenes are associated with the benzonium ion, could explain their reactivity as dienes of these compounds and the tendency to form cyclopentadienyl complexes [10]. Also is expected some changes in their magnetic properties as is observed from their calculated NICS values [22,25]. Thus, even very speculative at this moment, it’s clear that further elaborations and study on 16 could help to explain some of the unusual properties of metallabenzenes. This representation in 16, while it can appear non-intuitive, gives us a different perspective about the delocalization in metallabenzenes that is usually overlooked. We hypothesize that first and second row metallabenzenes tend to have a large benzonium ion component in comparison with their third row counterparts.
The dichotomy of 16 is related to the ability of the metal to donate a pair of π electron as in the cationic carbyne CH+ or their inability like in CH2 2+. There are a number of theoretical works about the electronic structure of metallabenzenes (see for instance [23,27]). These works reveal some differences in the π frontier molecular orbitals when compared with their aromatic analog, thus it’s clear that the pattern of bonding in these compounds are more elaborate that a simple isolobal replacement of a CH+ organic fragment. A reasonable indication of the degree of the two bonding pattern is the structural symmetry and the bond angles between the ligands around the metal. Since this dichotomy is present in most metallabenzenes, these molecules present a sort of bipolarity disorder, that manifest itself depending on the chemical environment.
Our hypothesis could be extended to 16-electrons metallabenzenes like the recent synthesized iridabenzene in which two phosphines of 1 were replaced by chlorines [39]. This last iridabenzene has an Ir(III) trigonal bipyramidal structure which is conceivable by starting from a 16-electron d6-ML5 to reach a structure analogous to benzonium ion. At this moment, this iridabenzene is one of the few 16-electrons compounds of this class. The importance of the hypothesis 16 is its capability to further elaborations on the basis of the combination of the properties of two extreme situations in an attempt to explain the unusual character of metallabenzenes. Also this approach to metallabenzenes can be extended to metallapentadienes and metallabutadienes.

