











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. Introduction
The use of accelerators to improve scientific computing performance is not exclusive of recent years. Their early application in scientific code can be traced back to the beginning of the 1980s, when a floating point accelerator was implemented in computers [3]. Recently, Graphic Processing Units (GPUs) have attracted a lot of attention, as can be seen in their extensive use in the high performance computing field.
Early use of GPUs was precluded by their inherent programming complexity, which relied on either OpenGL or DirectX graphic programming languages. This issue limited general purpose computation on GPUs and circumventing this limitation was the motivation for additional efforts [4. However, the release of NVIDIA’s compute unified device architecture (CUDA [5]) provided a high level abstraction model through the incorporation of relatively simple extensions of the standard C language, which permitted the development of libraries that are useful for common problems in quantum chemistry and solid state physics, such as Fourier transforms (CUFFT [6]) and linear algebra (cuBLAS [7]).
The success of this model is evident: since the CUDA release, a variety of codes have been developed for molecular dynamics applications [8,9], astrophysics simulations [10] and electronic structure methods. Within the last mentioned applications, special efforts have been made concerning the GPU implementation of Hartree-Fock (HF) [11,12], evaluation of electron repulsion integrals [13,14], density functional theory [15], geometry optimization [16], solvation models [17], resolution of the identity MP2 (RI-MP2) [2,18], coupled-cluster theory (CC) [19,20,21] and quantum Monte-Carlo [22]. In addition, popular electronic structure codes have adopted hybrid CPU/GPU schemes in order to speedup calculations, for instance GAMESS [23], NWChem 24], TeraChem [25] and Q-Chem [26].
Our work constitutes an additional effort to extend the scope of GPU applications in electronic structure methods. We are interested in the GPU implementation of the so-called scaled opposite-spin second order Møller-Plesset theory (SOS-MP2). This is a simplified and economical treatment of electronic correlation energy calculations [1]. In this approach, only the α-β component of MP2 energy is calculated and scaled by an empirical factor (which turns out to be 1.3), which yields statistically improved energies and derivative properties over the conventional MP2 method. In addition, the introduction of the Resolution of the Identity (RI) approximation, and a Laplace transform results in an improved method without any fifth order computational steps, in contrast with the original MP2 formulation.
A similar effort has been carried out recently by the introduction of a new SOS-MP2 algorithm by Maurer et al. [27] in which they reduce the scaling by modifying the rate determining step in such a way that it is efficiently evaluated in a GPU without using any GPU-based linear algebra library. However, we believe that our present work is still relevant as it complements the one initiated by Aspuru-Guzik and co-workers [2], by adding the non-blocking feature to the set of routines that constitutes the library in development. This has the advantage of reducing the programming effort in the addition of new matrix-multiplications related algorithms, at which the programmer could simply call a single function that automatically carries out the whole process of the non-blocking calculation.
In this work, the main details concerning the GPU implementation of the most computational intensive part of the SOS-MP2 algorithm of Q-Chem, are exposed. This article is organized as follows: in the Theory and Implementation section, the theoretical basis of the SOS-MP2 algorithm are presented and the details of the implementation process and the use of asynchronous calls in CUDA as an option to improve performance are described. In the Results section, the most important results concerning the process of benchmarking are discussed. In the Discussion section we comment on the speedups observed by the use of this GPU implementation. Finally in the Conclusions section we summarize our results.
2. Theory and Implementation
2.1. SOS-MP2 method
The theoretical basis of the SOS-MP2 method was reported by Head-Gordon and co-workers [1]. In this method, the energy is expressed as a series of matrix multiplications (within a discrete quadrature that involves Q points), according to:
 (1)
(1)
Here, the major computational task consists in the construction of the X matrix (or matrices, if open shell), which is defined in terms of three-center bielectronic integrals:
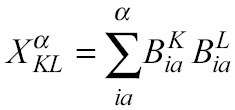 (2)
(2)
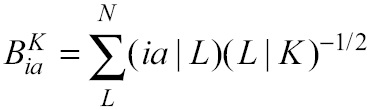 (3)
(3)
In Eq. 2,
 (4)
(4)
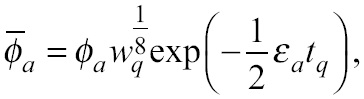 (5)
(5)
are a consequence of the numerical integration of
 (6)
(6)
according to
 (7)
(7)
Here,
In Q-Chem, for each quadrature point q, the X matrix (or matrices, if open shell) is constructed through the evaluation of Eq. 1. This step is fourth order in molecular size.
In actual Q-Chem SOS-MP2 calculations, the algorithm is timed in the six main steps that are illustrated in Figure 1, namely 1) construction and inversion of the (P | Q) matrix, 2) construction of the (ia | P) matrix, 3) construction of

Figure 1 Pseudocode showing the main steps in the SOS-MP2 energy evaluation algorithm in Q-Chem. In this scheme Q denotes the number of points in the numerical quadrature introduced in Equation 7. In ac tual calculations, Q = 7.
It is worth noting that the SOS-MP2 method preserves the size-consistency property given that the OS component of the MP2 energy conserves it, even though, it is possible that due to its approximate nature the size-consistency property might brake down in unexpected situations. Some physical deficiencies are known: firstly, it underestimates the correlation energy in the limit of long separation, due to the fact that the OS components and the same-spin (SS) ones are equal in this limit; and, secondly, it can overestimate the correlation energy in systems where the SS component is small [1].
2.2. Implementation
During the development of GPU applications, memory optimizations are fundamental to increase performance, thus a second goal is to maximize bandwidth in data transfer. This is commonly done through minimizing CPU-GPU communication, which can be achieved by batching many small transfers into a larger one or enhancing the bandwidth between the host and the device by taking advantage of the non-blocking data transfers capabilities available in modern GPUs [5]. The second option is realized using page-locked (or pinned) memory together with the so-called streams, which are sequences of commands (possibly issued by different host threads) that are executed in the order issued. In contrast with common blocking data transfers, the non-blocking version allows the programmer to issue several memory copies and kernel executions in different streams at a time, so that the operations can be interleaved and overlapped, which translates into a greater occupancy of the device memory (See Fig. 2).

Figure 2 Schematic timeline of asynchronous data transfer in the non-blocking developed version. This approach enables the program mer to overlap communication with computation time by issuing data transfers and kernel executions in different streams (streams are repre sented with the same color).
Data parallelism (which focuses on distributing the data across different parallel computing nodes) is common in high performance computing (HPC) oriented to scientific applications, an example of this model is the parallel programming “Single Instruction Multiple Data” of CUDA. Since the onboard memory of GPUs is a finite resource and the amount of data to be processed in HPC fairly exceeds this limit, it is usual to process chunks of the whole data set by the GPU in the most demanding tasks. However, the disadvantage of this scheme is that the number of data transfers that have to be issued is high for large data sets. Although in this case the communication host-device overhead can be reduced, when the algorithm allows it, by reducing data transfers (which can be done by batching several chunks into a larger one and executing the kernel sequentially over segments of the buffer). Nevertheless, in the GPU implementation presented here, we adopted a non-blocking model primarily because it permits to hide memory copy more effectively by overlapping CPU-GPU communication (and viceversa, i. e. GPU-CPU) with kernel execution. Additionally, the bandwidth is enhanced by using pinned memory, and different sets of data can be processed simultaneously when the device has this capability.
In order to show that the most demanding step of the SOS-MP2 algorithm corresponds to the one associated with Eq. 1, we carried out a benchmark to analyze the execution times of different steps involved in the SOS-MP2 algorithm, considering linear alkanes and employing the cc-pVDZ/rimp2-cc-pVDZ basis set. The results are summarized in Figure 3.

Figure 3 Comparison of the wall-time required by the algorithm steps of the SOS-MP2 energy evaluation, for linear hydrocarbons. The Dun ning cc-pVDZ basis and the rimp2-cc-pVDZ auxiliary basis set were used. Only the stages that account for the major percentages of total wall-time are shown: construction of three-center integrals (ia|P), con struction of
From these results, it is noted that the wall-time of the energy evaluation is dominated by the fifth step of the algorithm, which correspond to the calculation of the X KL α coefficients. Since it has been shown that this is the most expensive step in the algorithm, we investigated the possibility of achieving a speedup based on the use of Graphical Processing Units by working exclusively on this step.
Firstly, and in order to set goals regarding the speedup that can be achieved parallelizing the section of the code corresponding to the fifth step, we analyzed the trend in the percentage of the total wall-time SOS-MP2 energy evaluation spent in that section as the system size increases. This trend is illustrated in Figure 4.

Figure 4 Percentage of total wall-time required by the SOS-MP2 en ergy evaluation algorithm in the construction of
From these results, we noted that at most, step 5 represents approximately 82% of elapsed time, and the trend observed seems to prevail for bigger systems. Considering this proportion as parallelizable, and assuming that the 18% of elapsed time left is sequential, we can set the expected upper-bound speedup by employing Amdahl’s law [5]
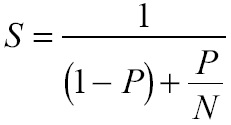 (8)
(8)
Where P is the fraction of the total serial execution time taken by the portion of code that can be parallelized and N is the number of processors where the parallel portion of the code runs. By using Eq. 8, we find that the maximum speedup that can be achieved assuming strong scaling is 5.6.
Equation 1 corresponds to the fifth step of the algorithm. In Q-Chem this is evaluated within a loop over active occupied molecular orbitals. If N is the number of auxiliary orbitals and v the number of active virtual orbitals, first a matrix of size v × N is read from a temporal file and it is multiplied by its transpose afterwards. Then, this result is augmented into a N × N matrix X. In order to use the GPU more effectively, we implemented a batch scheme to group K matrices together in such a way that each matrix is processed by an independent stream.
3. Results
The most intensive part of the algorithm, as shown above, corresponds to matrix multiplications. In order to assess the performance of this operation with two different CUDA approaches (namely non-blocking and blocking communication versions), we developed two different code toy models. Both models process 100 random matrices, multiplying each of them by its transpose and augmenting the result into a final matrix. The difference between them resides in that one uses asynchronous communication (non-blocking version) and the other uses the common communication routines (blocking version). These test codes were written with CUDA version 6.0 (CUDA driver version 6.0) and were compared with the performance achieved by the Intel® MKL library (Fig. 5). The calculations were carried out on Intel Xeon® processors at 2.40 GHz, on a node with the Red Hat Enterprise Linux OS, release 6.5. In the GPU calculations, a Tesla K40C GPU was employed. From the results obtained (Fig. 5), it is worth noting that the reduction in performance due to PCI bus latency is minimal in the case of small matrices, when the non-blocking communication approach is employed (as a result of the computing-communication overlap), and the maximum observed speedup with respect to CPU timings is approximately 6 % greater that the maximum speedup using blocking communication.

Figure 5 Performance comparison between the two developed CUDA versions for data processing. Speedup=CPU elapsed time/GPU elapsed time.
It is worth mentioning that the performance achieved using the MPI model is approximately the same as that achieved when six CPUs are used in conjunction with the use of a GPU as a coprocessor. However, as previous work shows [28], one of the advantages of GPU computing over a multicore approach, is the low energy amount per floating point operation in numerical computations that involve BLAS level 3 operations (matrix-matrix operations). Although a mixed MPI-CUDA scheme can be proposed to take advantage of modern clusters with several GPUs and CPU cores, we preferred to employ the CUDA scheme in the current study, given the availability of a matrix multiplication library in the Q-Chem code, and considering that the new functions can be easily incorporated in a future MPI implementation.
For comparison and exploration purposes, the first approach used to introduce acceleration in the calculations was a simple substitution of the subroutine that carries out the matrix multiplication used during the construction of the
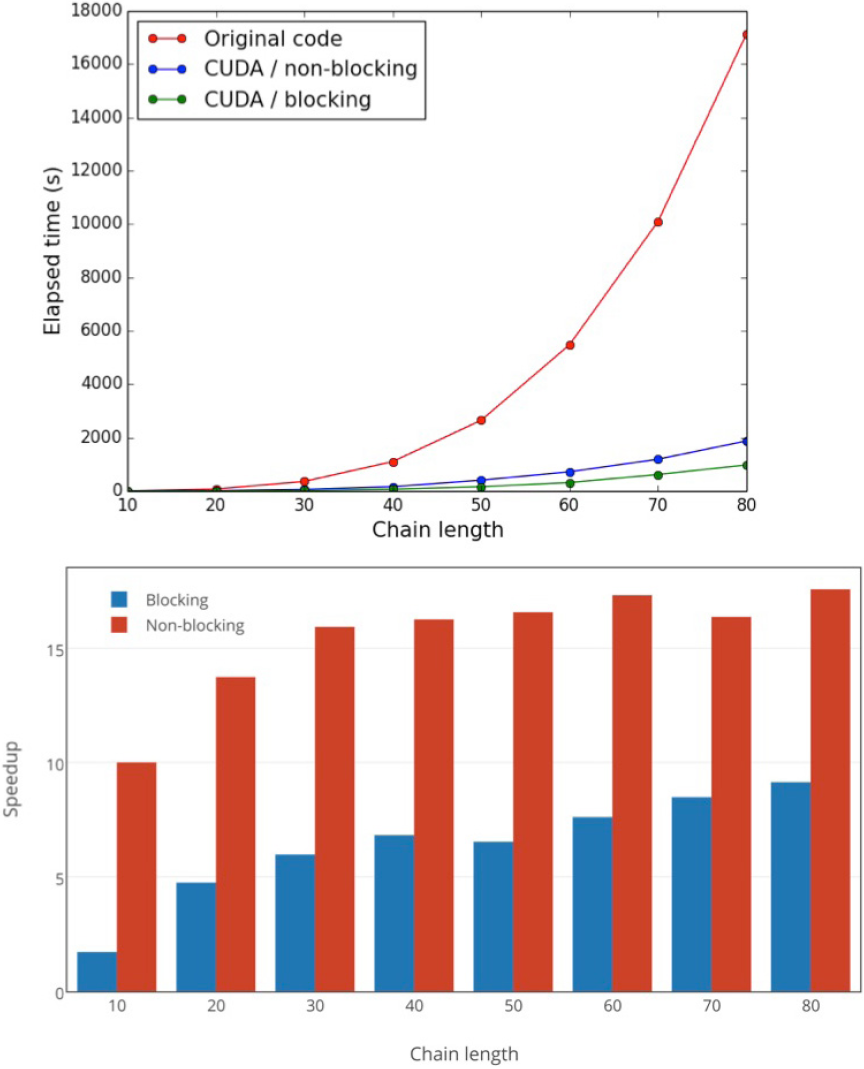
Figure 6 (a)Timing comparison between blocking and non-blocking versions, developed in this work, for the construction of
To introduce non-blocking communication and with the aim on concurrent execution of matrix multiplications, we proposed a batching scheme in which batches of K matrices are initialized and then are processed and multiplied by its transpose within a single call of a function, carrying out these tasks in different streams for each matrix. The value of K is set dynamically according to a threshold value which reflects the onboard GPU memory constraint. From the results illustrated in Figure 6, we note that this approach introduces a significant time improvement, with respect to the blocking version. However, this improvement seems to be less effective when the system size increases, as a consequence of the reduction in the percentage of time invested in communications.
In order to include all these advantages in Q-Chem, a series of subroutines were implemented within the code responsible of the computation of the SOS-MP2 energies. These subroutines accomplish the streams setup, the matrix multiplication using CUBLAS in a concurrent and batched fashion, and the shutting down of the mentioned streams. Furthermore, to take into account the possibility to process big matrices that do not fit the GPU on-board memory, we employed the matrix multiplication library developed by Aspuru-Guzik and co-workers 2] which has the capability to chop big matrices and process the matrix multiplication by pieces. The modified algorithm is described in more detail in Figure 7. The speedups achieved with the implementation of these subroutines are shown in Figure 8. To avoid possible fluctuations in the speedups data due to changes in processing time of the different stages of the calculations, we considered that the only variable in the calculation wall-time was the time spent on step 5, between the GPU and CPU version. This was done since we noticed that the speedup calculations based on the GPU and CPU raw data gave rise to spurious speedups above the maximum theoretical speedup for the biggest systems.

Figure 7 Pseudocode showing the main steps of the proposed algo rithm to build the X matrix in SOS-MP2 calculations. As in Figure 1, Q denotes the number of points in the quadrature for the evaluation of Eq. 7.

Figure 8 Speedups achieved in the calculation of the correlation energy of SOS-MP2 algorithm after the implementation of the asynchronous CUDA approach described in the main text. The basis set employed is cc-pVDZ.
We tested also for the numerical precision of results obtained on a GPU as coprocessor as they compare to results run on a CPU. As expected for double-precision calculations on both CPU and GPU, results are equal, except for one alkane whose correlation energy is within a margin of less than 0.1 kcal/mol. Since there was only one case of slight discrepancy between GPU and CPU results, we believe that the error source is not algorithmic but due to an error in data processing.
4. Discussion
We have found that the concurrent capacity supported by the majority of GPUs introduces an important enhancement in the execution of the most demanding step of the SOS-MP2 algorithm. On the other hand, given that the speedup in this algorithm has an upper bound of 5.6, the increase in performance compared with the blocking version of the code seems small (see Figure 6), yet it is noteworthy that the speedup achieved constitutes approximately 87% of that maximum value.
On the other hand, it should be mentioned that the main drawback of this method is that pinned memory is a scarce resource which means that an excessive use of this kind of memory can reduce the overall system performance, that limits its use in very big systems. Unfortunately it is difficult to know how much page-locked memory allocation can interfere with performance, given that it depends on the operative system and other OS applications that compete for resources. However, we think that this problem can be ameliorated with the use of appropriate queue systems that limit the amount of resources allocatable by users, which is the case in modern clusters oriented to HPC.
5. Conclusions
In this paper we have evaluated the most computationally intensive part of the SOS-MP2 algorithm for the calculation of the correlation energy with particular application to its execution within the Q-Chem suite. We built on the asynchronous CPU-GPU communication capability of modern GPUs to increase bandwidth and performance. These capabilities were tested on the calculation of a set of linear alkanes from 10 to 80 carbon atoms, where we were able to find an almost three-fold increase in the speed of the computation of the correlation energy. While these results are slightly different depending on the particular GPU employed, considerable speedups are present in all cases. As implementation of the proposed code is simple, use of mixed GPU-CPU schemes like the present one are recommended.

