











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
The 1,8-naphthyridone derivatives have received significant attention due to their exceptionally broad spectrum of biological activities. The first 1,8-naphthyridone group was nalidixic acid used as a synthetic quinolone antibiotic [1,2]. However, this quinolone showed activity only against Gram-negative microorganisms. Subsequent structural modifications of this quinolone led to a wide range of biological properties established them as potent scaffolds in therapeutic and medicinal research. The broad spectrum of activities includes antibacterial [3,4], antitumor [5-9], antiviral [10,11] and some antimycobacterial activities [12,13] as well as anti-inflammatory activity [14-17]. The structural features of the more potent, newer generation quinolones such as tosufloxacin, trovafloxacin, enoxacin and gemifloxacin (Figure 1) include C-6 fluorine and a C-7 nitrogen-containing heterocycle including piperazine or pyrrolidine into the 1,8-naphthyridone group [3,4,16].
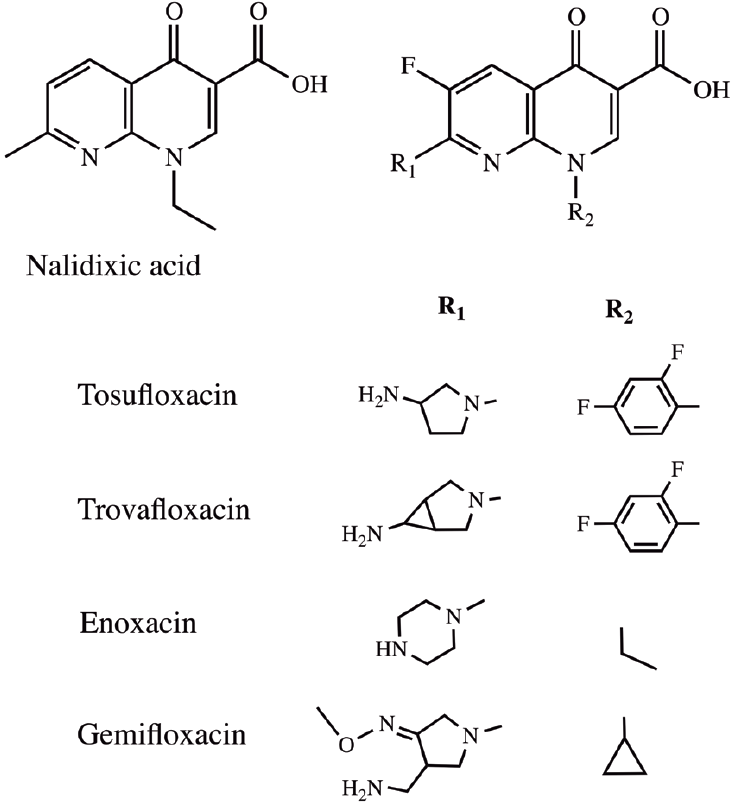
In spite of a significant number of fluoroquinolones being approved for treatment of bacterial infections [18]; there is still need for developing new derivatives that could overcome the emerging problem represented by bacterial chemoresistance. Furthermore, some antibacterial quinolones have important significant side effects. For example, trovafloxacin was recently removed due to its liver toxicity [19].
Numerous synthetic methods have been reported for the preparation of quinolones under conventional conditions; however, these methods describe long reaction times, low yields and elaborated work-up procedures. In this paper, we describe a fast microwave-assisted synthesis for the preparation of ethyl 7-chloro-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylate reported in the literature [5,6,20,21].
Results and Discussion
The 1,8-naphthyridine ring can be prepared by two-step synthesis of 3 as showed on Scheme 1. First, 2,6-dichloronicotinic acid (1) is reacted with 1,1-carbonyldiimidazol (CDI) to give an imidazolide intermediate at room temperature and 2-2.5 h, according to the literature [5, 6, 21]. Which reacts with ethyl malonate potassium, MgCl2 and triethylamine to generate ethyl 3-(2,6-dichloropyridin-3-yl)-3-oxopropanoate (2) using intervals of room temperature to 600C and 1-15 h as reactions conditions, reaching 59-93% yield. Nevertheless, these conditions were improved using microwave radiation, decreasing the reaction time to 5 min for the synthesis of imidazolide intermediate, the rest of the reaction was performed according to a previous report described in the literature with a 93% yield.
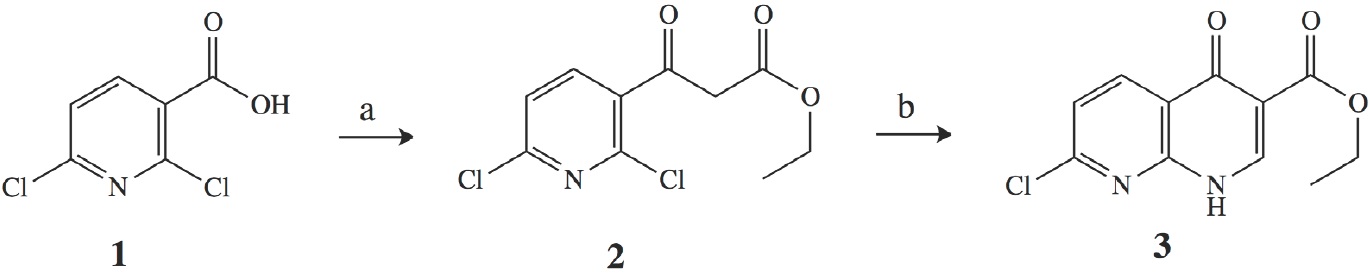
(a)1) CDI, THF/CH3CN, MW, 5 min; 2) EtOCOCH2COOK, MgCI2, Et3N, RT; (b)1) CH(OEt)3, AC20, MW, 3.6 min, 2)CeCI3,NH3 or (NH4)2CO3/1,4-dioxane, MW, 15 min
Scheme 1 Microwave-assisted synthesis of ethyl 7-chloro-4-oxo-l,4-dihydro-1,8-naphthyridine-3-carboxylate.
Then, a usual route of synthesis proposes that molecule 2 reacts with acetic anhydride and triethyl orthoformate, to generate an intermediate vinyl ether in 55 to 90% yield, using a nitrogen atmosphere and temperatures between 130-140 ºC. Following by the addition of an amine or amine substituted at room temperature for 1-5 h or overnight, resulting 55 to 90% yield. After this, the synthesized intermediate proceeds to cyclization reaction using 50-600C and time lapse of 1-3 h, with 63-92% yield of compound 3. A pathway with three-step synthesis that can be easily converted into one-pot synthesis. The molecule 2 reacts with acetic anhydride and triethyl orthoformate under MW conditions (70W, 140 ºC, 3.6 min) to yield an intermediate vinyl ether in which CeCl3 and either NH3 or (NH4)2CO3 was added to produce a vinyl amine using 100W and 100 ºC that allowed easily undergoes cyclization at the same time. The crude product obtained, was purified by a simple column chromatography using hexane to obtain the 1,8-naphythyridone product 3. The yields using NH3 or (NH4)2CO3 were 73% and 75% respectively.
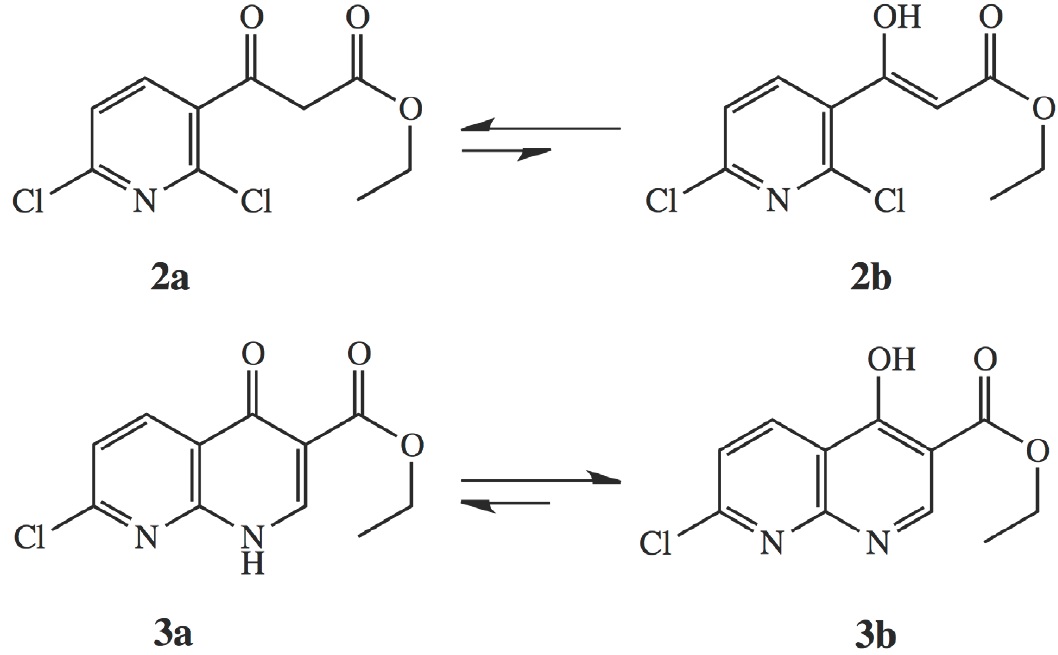
Compounds 2 and 3 were confirmed by 1H NMR and FT-IR spectroscopy [19, both presented a keto-enol tautomeric equilibrium as shown in Scheme 2. Tautomer 2a had a CH2 signal (4.09 ppm) while 2b gave a broad OH signal (12.54 ppm) and a vinyl H signal (5.72 ppm). In the case of 3a, a vinyl H signal (8.53 ppm) and a broad NH signal (1.59 ppm) were observed while 3b, an OH signal (2.57 ppm) was obtained. According to an extensive aromatic system and intramolecular hydrogen bond, the enol tautomer 3b was strongly favored. Furthermore, the FT-IR spectra of 2 and 3 showed absorbance bands corresponding to the presence of O-H alcohol, C=O enolic ester, as well as a C=O ester and a ketone.
Experimental
The reactions were carried out using a CEM monomode MW synthesizer with a CEM Discover System workstation and monitored by thin-layer chromatography (TLC) using silica gel 60 F254 plates, eluting with hexane/ethylacetate. FT-IR spectra were obtained in KBr pellets on a Fourier Nicolet (iS10 FT-IR) spectrometer. NMR spectra were recorded on a Varian Mercury Plus 400 MHz spectrometer. Mass spectra were done on a JEOL MStation JMS-700 apparatus.
Procedure and spectral data
Ethyl 3-(2,6-dichloropyridin-3-yl)-3-oxopropanoate (2a) and ethyl 3-(2,6-dichloropyridin-3-yl)-3-hydroxyacrylate (2b)
To a solution of 2,6-dichloronicotinic acid (0.20g, 1.04 mmol) in THF-CH3CN (1:1, 3.0 mL) was added 1,1-carbonyldiimidazole (0.19 g, 1.17 mmol). The resulting mixture was stirred and heated using MW (5 Watts power at 50ºC for 5 min). This crude imidazolide solution was used without purification in the following step. To a solution of ethyl malonate potassium salt (0.19 g, 1.09 mmol) in CH3CN (2.50 mL) was added dropwise a mixture of MgCl2 (0.14 g, 1.52 mmol) and Et3N (1.50 mL) under ice cooling. After the mixture was stirred at room temperature for 5 h, the imidazolide was added. The reaction mixture was stirred at room temperature for 15 h, poured onto ice water (5 mL) resulting in a pH about 11, so we acidified this mixture using concentrated HCl to pH = 6. The crude product was extracted with AcOEt, dried with Na2SO4 and concentrated under vacuum to give product 2, obtained as a yellow oil (93% yield). The product consisted of a mixture of the ketone 2a and the enol 2b tautomers (confirmed by spectroscopic methods) [6.
IR (KBr) nmax 3133 (C-H aromatic), 2984, 2936 (C-H aliphatic), 1761 (C=O ester), 1736 (C=O ketone), 1631 (C=O ester enolic), 1573 (C=C aromatic), 1541 (C=N aromatic), 1385 and 1341 (C-N aromatic), 1282, 1242 and 1141 (C-O ester and alcohol), 1095 and 1020 (C-Cl aromatic), 843 and 772 (C-H aromatic out of plane) cm-1. MS, m/z (%): 262.0 (M+, 21), 216 (13), 167 (32), 149 (85), 136 (46).
(2a) 1H NMR (CDCl3 400 MHz) δ 1.26 (t, 3 H, J = 7.20 Hz), 4.09 (s, 2 H), 4.20 (q, 2 H, J =7.20 Hz), 7.40 (d, 1 H, J = 8.0 Hz), 7.98 (d, 1 H, J = 8.0 Hz). 13C NMR (CDCl3 400 MHz) δ 192.41 (C=O ketone), 172.39 (C=O) ester, 152.93, 141.69, 141.41, 140.85 and 123.04, (aromatic C), 60.38, 48.68 (CH2) and 14.01 (CH3).
(2b) 1H NMR (CDCl3 400 MHz) δ 1.35 (t, 3 H, J = 7.20 Hz), 4.29 (q, 2 H, J = 7.20 Hz), 5.72 (s, 1 H), 7.36 (d, 1 H, J = 8.0 Hz), 7.94 (d, 1 H, J = 8.0 Hz), 12.54 (s, 1 H). 13C NMR (CDCl3 400 MHz) δ 171.16 (C=O) ester, 166.57 (C-OH alcohol), 152.94, 141.69, 141.41, 132.47 and 123.04 (aromatic C), 123.46 (vinyl C), 61.78 (CH2) and 14.16 (CH3).
Ethyl 7-chloro-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylate (3a) and ethyl 7-chloro-4-hydroxy-1,8-naphthyridine-3-carboxylate (3b)
A mixture of 2 (0.40 g, 1.50 mmol) and acetic anhydride (0.36 ml, 2.25 mmol) was heated using MW at 70 Watts until it reached 140ºC. Then triethyl ortoformate was added (0.38 mL, 3.75 mmol) stirring under MW heating (70 W, 140ºC) for 3.60 min. The reaction mixture changed from yellow to an intense red color. AcOEt and CH3CO2H were distilled off under reduced pressure. The resulting crude product was mixed with CeCl3.7H2O (5.60 mg) and stirred for 10 min. Then, a solution of either NH3 or (NH4)2CO3 (2.50 mmol) in 5.10 mL of 1,4-dioxane was added in three fractions to the mixture under open vessel MW at 100 Watts until it reached 100ºC for 15 min. The crude product was purified by column chromatography using hexane as the mobile phase to obtain 3 as colorless solid in a mixture of ketone 3a and enol 3b tautomers with m.p. 283-286ºC [20. 73% yield was achieved by using NH3, while in the case of (NH4)2CO3 the yield was 75%. IR (KBr) nmax 3423 (O-H alcohol), 3071 (C-H aromatic), 2986, 2926 and 2852 (C-H aliphatic), 1723 (C=O ester), 1664 (C=O ketone), 1623 (C=O ester enolic), 1594 (C=C aromatic), 1550 (C=N aromatic), 1406 (C-N amine), 1385 (C-N aromatic), 1289, 1219 and 1130 (C-O ester and alcohol), 1073 and1016 (C-Cl aromatic), 796 (C-H aromatic out of plane) cm-1. MS, m/z (%): 222 [(M-CO-H2)+, 9], 220 (16), 167 (20), 154 (100), 149 (79), 137 (55).
(3a) 1H NMR (CD3COCD3 400 MHz) δ 1.41 (3 H, t, J = 9 Hz), 1.59 (1 H, bs, NH), 4.42 (2 H, q, J = 9 Hz), 7.36 (1 H, d, J = 10 Hz), 8.15 (1 H, d, J = 10 Hz), 8.53 (1 H, s). 13C NMR (CDCl3 400 MHz) δ 173.86 (C=O ketone), 164.03 (C=O ester), 159.53, 155.80, 141.96, 123.42 and 120.56 (aromatic C), 142.23 and 116.80 (vinyl C), 62.32 (CH2) and 14.10 (CH3).
(3b) 1H NMR (CD3COCD3 400 MHz) δ 1.40 (3 H, t, J = 9 Hz), 2.57 (1 H, s, OH), 4.42 (2 H, q, J = 9 Hz), 7.27 (1 H, s), 7.46 (1 H, d, J = 10 Hz), 8.52 (1H, d, J = 10 Hz). 13C NMR (CDCl3 400 MHz) δ 163.46 (C=O ester), 163.08 (C-OH alcohol), 156.67, 154.95, 148.29, 139.06, 122.78, 111.17 and 110.92 (aromatic C), 62.12 (CH2) and 14.15 (CH3).
Conclusion
An improved Grohe-Heitzer methodology [21-24 was developed by the use of microwave energy reducing 2.5 h to 5 min for the synthesis of the intermediate imidazolide. Which was employed in the generation of ethyl 3-(2,6-dichloropyridin-3-yl)-3-oxopropanoate with 93% yield, while the preparation of ethyl 7-chloro-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylate was made as one-pot reaction with 73% yield resulting in shorter reaction times and cleaner products. This useful alternative procedure could be helpful to prepare the skeleton of 1,8-naphthyridone agents.

