




![Synthesis and Characterization of Three New di-n-butyl [bis (alkyl-aminopropionic acid)]tin (IV)](/img/pt/next.gif)






 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink1. Introduction
The benefit of generics usage in reducing healthcare expenditure has been proven worldwide. The manufacturing and usage of generic medicines continues to increase and has been well recognized for a long time [1, 2]. The development of excipients or preservatives used in the manufacture of generics may vary from one company to another, and has given rise to numerous combinations. Due to this fact, some active ingredients are analyzed in new pharmaceutical matrices where there is no test method reported. This issue has presented a new challenge to the analytical chemistry field. This is the case of thimerosal (TMS), a mercurial organic compound that has been widely used as an antimicrobial agent and a preservative in sterile pharmaceutical formulations, including multi-dose vaccines, topical antiseptic solutions, intravenous preparations and ophthalmic solutions [3,4] and also in topical pharmaceutical creams. Over time, TMS has been reported as potentially dangerous to humans because it can change into methylmercury, which is a type of toxic mercury that can easily penetrate biological membranes and causes bio-accumulation. Hence, stability studies have also been performed [5-7]. Safety concerns and potential risks to human health, especially in children have originated a decrease of its use in pharmaceutics [3, 8, 9], where content usually varies from 0.001 to 0.15 %. In topical creams, TMS concentration is usually 0.1% w/w[4]. Consequently, sensitive and specific analytical methods for control of TMS immersed in different types of matrices remains of paramount interest.
Analytical methods of preservatives in in parenteral products have been reviewed [10]. Several methods have been published for the determination of TMS in different kind of sample matrices. Most of them are focused on its application in sterile pharmaceutical formulations. Thus, sensitivity methods based on total Hg quantification, involving mercury vapor generation, has been performed in vaccines (PVG-OES) [11] (DBD-PIV-AFS) [12] or in ophthalmic solutions by flow injection (FI) coupled with UV/microwave-assisted photochemical (CVG-AFS) detection [13]. Direct determination of TMS, using high performance liquid chromatography (HPLC) has also been developed in vaccines [14-16] or ophthalmic solutions [17-21] . Thimerosal degradation in aqueous medium may influence the preservative action. Therefore, stability studies in this medium have been performed [22, 23]. Likewise, HPLC coupled to an atomic fluorescence spectroscopy detector, has been performed in effluent and river waters by the pharmaceutical industry [24]. In addition, electrochemical methods have been described [25-27] as well as colorimetry [19]. In addition, the USP 39 reported TMS determination in topical solutions, tincture and topical aerosol [28]. Other methods of TMS analysis include early platforms such as inductively coupled plasma (ICP) and mass spectrometry (MS) [28,29]; however, both have the drawback of expensive equipment.
The only developed HPLC-UV methods [15, 16, 22] have been applied in vaccines and ophthalmic solutions. Particularly in topical creams, the quantification of TMS is quite difficult due to the oily nature of its matrix, low content and the instability properties of TMS [22]. There is no analytical method developed for quantification of TMS in topical creams, to our knowledge. A full validation of a HPLC-UV method for TMS determination in topical creams is proposed in this paper according to the ICH guidelines. The current work presents a simple sample treatment and involves the quantitative extraction of TMS from pharmaceutical creams. The method was applied in topical pharmaceutical creams containing 0.01% of fluocinolone acetonide (FLA) as the active compound.
2. Materials and Methods
2.1 Chemicals and Reagents
Methanol HPLC grade and potassium phosphate monobasic were obtained from J.T. Baker. Phosphoric acid from Reactivos Química Meyer (DF, México), and sodium hydroxide were purchased from Macron Fine Chemicals (PA, USA), both were analytical grade. Ultrapure deionized water from Milli-Q system (Millipore, Bedford, MA, USA) was used throughout. Mobile phases were filtered through 0.45 µm membranes (Millipore) and degassed prior to use. TMS secondary standard (98.6%), topical cream containing FLA and TMS. Placebos and topical cream were prepared and provided by Productos Farmacéuticos S.A. de C.V. (Aguascalientes, Mexico) which manufactures and sells this topical cream in México. 0.45 µm Nylon, GHP, PVDF acrodiscs (Millipore) were for sample preparation.
2.2 Chromatographic system and conditions
The liquid chromatography system employed was a Waters HPLC (Waters, USA) equipped with a Millennium Pro (Version 2002) chromatography manager for data acquisition and processing. This was equipped with a Waters 600 pump, a UV-Vis photodiode array detector (Model 996), and a Waters 717 automatic sample injector.
The optimized method was performed on a C18 reverse phase column (Symmetry®, 250 x 4.6 mm I.D., 5 µm, Waters, USA) equipped with a guard column of the same packing material. The mobile phase consisted of methanol and phosphate buffer 0.05 M adjusted to pH 2.5 (70:30 v/v). TMS was monitored at a wavelength of 218 nm with a mobile phase flow rate of 0.7 mL/min. The injection volume was 60 µl and total running time of 12 minutes.
2.3 Sample preparation
Topical cream and placebo were provided by Productos Farmacéuticos S.A. de C.V. (México). Samples were prepared from placebos whose formulation was identical to comercial cream. These placebos were spiked with known concentrations of TMS, as follows: approximately 1 g of topical cream containing the equivalent amount of 20 µg of TMS was carefully weighted in a 50-mL low-actinic Erlenmeyer flask. A volume of 7 mL of 0.2M phosphate buffer pH 5.5 was added, and placed in an ultrasound bath at 45 °C for 30 min (System A). Afterwards, system A was stirred with a magnetic agitation for 20 min at 60 rpm. Later on, system A was carefully transferred to a 10-mL low-actinic volumetric flask and diluted with phosphate buffer to the mark (System B). System B was transferred into a tube to be centrifuged for 15 min at 3500 rpm. Supernatant fluid was removed and filtered through a 0.45 µm PVDF acrodisc. Samples were put into actinic vials for HPLC analysis. The final concentration was 2 µg/mL, approximately.
Thimerosal standard. The standard solution was prepared daily as follows: an equivalent quantity to 20 mg of TMS was weighted, dissolved and diluted with water to the mark in a 100-mL low-actinic volumetric flask (Stock solution). From the stock solution an aliquot of 5 mL was taken and diluted with phosphate buffer (pH 5.5, 0.2 M) to the mark in a 100-mL actinic volumetric flask (Solution A). A second dilution was performed taking an aliquot of 5 mL from Solution A and diluted with phosphate buffer to the mark in a 25-mL actinic volumetric flask. Resulting solution was 2 µg/mL approximately, (Solution B).
2.4 Validation Studies
Developed method was validated in agreement with ICH guidelines [31], with emphasis on system suitability, specificity, linearity and range, precision, accuracy, limit of quantification (LOQ) and detection (LOD), robustness and extracted sample stability. In particular, specificity was demonstrated by comparing chromatograms from samples containing TMS against placebo and standard. Moreover, peak purity test was carried out showing that chromatographic peak was attributable only to one component. System suitability was evaluated injecting six replicates of TMS standard solution at 100%. Linearity was estimated by assaying at least five levels of concentrations of TMS by triplicate, covering values ranges from 75 to 125% of mean value found in topical creams. Recovery was tested on spiked cream samples by comparing five concentrations in triplicate from 75 to 125% within the standard curve range. Within-day precision of the method was checked by injecting individual preparations of standards and samples in the midrange of the calibration curve (n=3) by different analyst. Inter-day precision was evaluated in the same way, but on a different day, with freshly prepared buffers and reagents (n=6). Accuracy was tested using six spiked samples prepared at the 100% TMS of linearity. LOQ and LOD were calculated as 10α/A and 3.3α/A, respectively where α is the standard deviation (SD) of B-intercept and A is the slope of calibration curve.
The robustness was determined by estimating the influence of deliberate changes in the analysis conditions associated with the quantification of TMS. Factors such as flow rate, proportions of mobile phase and injection volume were studied. Finally, the time after sample is prepared where TMS still stable and can be analyzed by HPLC was tested. TMS stability in solution was checked on three processed cream samples under different storage conditions; temperature, amount of light and time. Samples for stability in solution were analyzed on the preparation day and after 6, 12 and 36 hours of storage in darkness either at room temperature or at 4 °C in the fridge.
3. Results and discussion
The performance parameters of the proposed method were adequate for the detection and quantification of low concentrations of TMS from oily pharmaceutical forms whose active substance is FLA.
3.1 Method development
The critical step in the analysis TMS in topical creams was the sample preparation due to its oil nature and its low content. For achieving the maxima extraction of TMS, solvents such as water and chloroform, different pH´s buffer solutions, filter type, temperature, times, and velocities of ultrasonic bath and centrifuge, were investigated. The main purpose was to achieve a quantitative TMS recovery in pharmaceutical cream containing fluocinolone acetonide as the active substance. For this purpose, different extraction solvents were tested including water, ethanol, acetone, isopropanol and chloroform. Unfortunately, water and acetone produced a cloudy solution; isopropanol dissolved all sample components and chloroform showed a poor TMS recovery. When using etanol 95%, a TMS recovery about 71% was found. In order to increase it, water was used instead, adjusting the pH of 0.2M phosphate buffer above the pKa of TMS (pKa = 3.05) to favor its ionized form and therefore its solubility in this medium. The assayed pH values were 4.5, 5.5, 6.5, 7.5 and 8.5, being pH = 5.5 the best value to separate TMS from excipients of the formula with a recovery of 84%. Likewise, different acrodisc types (Nylon, GHP and PVDF) and magnetic stirring times were evaluated. The recoveries were increased using PVDF acrodisc and with 20 minutes stirring time (Table 1). Finally, in order to investigate the reproducibility of this process, six samples of topical cream prepared and analyzed. The results showed maximum average recovery of 100.4% (RSD 0.72). Reproducibility of this methodology proved it was suitable for sample treatment.
Table 1 Optimization of the percentage recovery as a function of used acrodisc type*

* Experimental procedure: Sample was mixed with the 10 mL of phosphate buffer (0.2M, pH 5.5) and placed in an ultrasound bath at 45 °C for 30 min., afterwards the system was magnetically stirred for the corresponding time at 60 rpm. The solution was centrifuged for 15 min at 3500 rpm and the aqueous phase was removed and filtered through the corresponding acrodisc type (0.45 µm).
3.2 Validation
Once chromatographic conditions and sample treatment were established, method validation was performed following ICH guidelines [31]. The results proved that validation parameters were adequate for the analysis of TMS in topical creams containing FLA (0.1 %) and agreed with ICH (Table 2).
Table 2 Summary of validation parameters for TMS in topical cream containing FLA (0.1%)*.

* A, slope (± standard error); B, intercept (± standard error); §: Area/Concentration (%RSD); &: Estimate Concentration/Real Concentration (%RSD).
** Peak area = A(TMS concentration) + B
*** Analyte recovery = A(Analyte spiked) +B
Suitability of the system showed a RSD % less than 2% and chromatographic performance parameters fully met the criteria of acceptance in accordance with ICH guidelines. The selectivity was proved by comparing the standard, sspiked sample and placebo chromatograms (Fig. 1). In the chromatogram of placebo, no peak was observed at the retention time of the TMS. In addition, the peak purity was verified using the DAD detector, indicating that the analytical signal detected was solely due to TMS. The standards and samples showed a good linear regression with determination coefficients greater than 0.99 and no bias was observed. Calibration (standard) curve was generated using 1.2, 1.4, 1.6, 1.8, 2.0 mg/mL. The confidence interval of the y-intercept included zero. Spiked samples ranged from 75 to 125 %. The RSD of slopes ranged between 1.3 and 1.8 %, which represents the good fit of individual points to the regression line for TMS in standards and samples. The confidence interval of the slope included the unity. Both, linearities of calibration curve and method, fulfilled the ICH criteria [32]. (Table 2). Method precision had RSD values on the first day of 1.5 and 2.1 % by two analysts while 1.6 and 1.4 % from both days for each analyst. For the accuracy of the method, RDS values were 1.5 and 2.1 % by two analysts on the first day and 1.6 and 1.4 % per analyst for two days. Therefore, precision results were agreed with validation requirements for inter- and intra-day analyst. Recovery for TMS was found approximately 99 % ±1.01 in the topical creams forms, which fulfilled the validation criteria. The LOQ and LOD values were 0.79 and 0.26 µg/mL, respectively, which proved high level of sensitivity of current method for TMS quantification.
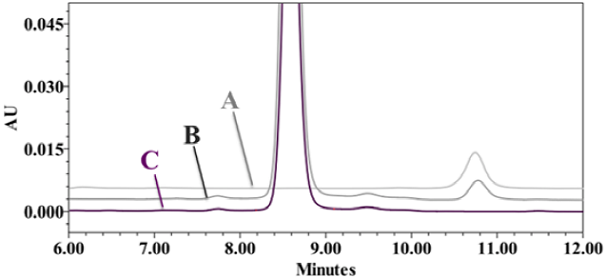
Figure 1 Selectivity of the analytical method. Chromatograms comparison. (A) Standard of Thimerosal (TMS, 2 µg/mL); (B) spiked placebo (FLA and TMS); (C) Placebo sample (FLA). Conditions: Symmetry® RP C18 column, methanol: phosphate buffer (pH 2.5, 0.05 M) 30:70 v/v, flow rate of 0.7 ml/min, UV detection at 218 nm, 60 µL of volume injection.
Table 3 shows the results of method robustness tests. Mean recovery values proved a satisfactory reproducibility of the method when slight changes in mobile phase composition, flow rate and injection volume are made. The RSD values minor than 2.0 % except for the highest level of methanol, showed a good precision of the method.
Table 3 Robustness results of the analytical method for TMS quantitation in topical creams containing FLA 0.1%

*Methanol: phosphate buffer (5mM, pH=2.5) (v/v); &Mean recovery = Average (%), C.I. =Confidence Interval.
In relation to stability TMS, after sample preparation, results showed that it kept stable for 12 hours under dark storage room temperature, or 36 hours at fridge temperature of 4 °C (Table 4) with RSD values less than 1 % for all the cases, except at room temperature for storage 36 hours, where RSD value was 3.5 %. The analytical method was carried out with extensive validation parameters as per ICH guidelines proving to be accurate, precise and robust.

3.3 Comparison of HPLC methods
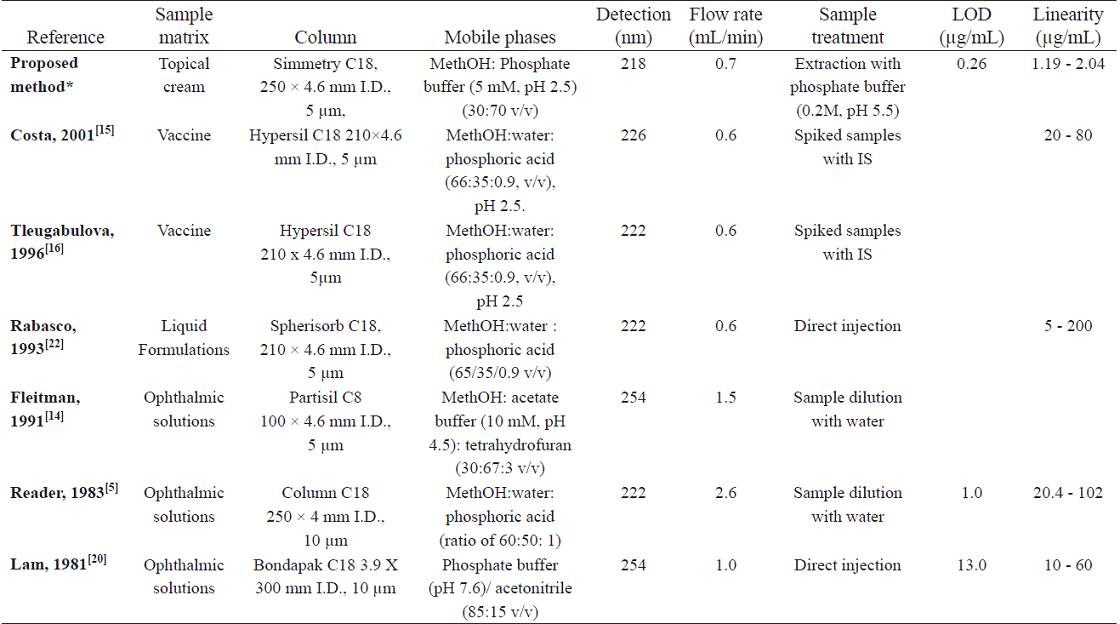
Up to now, all published methods were developed and applied rather in aqueous samples while in this study is presented the development of a method for a difficult matrix sample such as a topical cream. As well, the present method reached the lowest concentration limit of 1.2 µg/mL compared to the obtained by Rabasco, et. all [22] where it was 5 µg/mL. It is noteworthy, that the LOD and LOQ have not been reported, whereas the value observed in this method shows the good sensitivity of the analytical method. All reported methods, including ours, used the same type of column and mobile phase of similar composition (Table 5).
4. Conclusions
The present assay HPLC method was carried out with validation as per ICH guidelines. This validated method was demonstrated to be accurate, precise and robust. The method was found to be linear (R2=0.9957) within the analytical range of 1.2 a 2.02 µg/mL. A maximum recovery of TMS was achieved from pharmaceutical cream using 0.2 M Phosphates buffer at pH 5.5. In addition, the TMS was found to be stable under various processing and storage conditions, 12 hours at either room temperature or at 4°C. In summary, the developed and validated HPLC method that showed to be adequate for quantification of TMS in the routine analysis of pharmaceutical topical creams in a quality control laboratory.

