











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink1. Polypharmacology
Currently, the main paradigm in drug discovery is the development of target-specific inhibitors. This also implies molecules with high-fold potency and selectivity towards one isoform. This mainstream view has its origins in the so-called “magic bullet” as enunciated by Paul Ehrlich over 150 years ago. Indeed, such concept was engraved in the mind of many health professionals and researchers as the top achievement in drug discovery. However, as years came by, this has proven to be a disappointment mainly because of the off-target responses, which may involve toxicological concerns or side-effects. For example, considering the wide array of enzymatic systems, classes and isoforms identified in biology it is no wonder that many target-specific agents had been developed via trial-and-error approaches [1].
Recent statistics show that pharmaceutical industry is struggling as many promising drugs fail during the early stages of drug development along with the associated significant economic disadvantages [2]. This shows we have reached an impasse: just between 1996 and 2001 a large number of drugs were withdrawn from the market because of similar reasons [3]. Furthermore, even selective drugs are not exempt of drug-drug interactions which also represent a challenge, especially for chronic therapies. After reaching this point, we must ask ourselves if this mainstream view needs refinement or a drastic change of perspective. So if target-based drug discovery has not lived up to expectations, what choices are left? What if the so-called side effects are not “failures” after all? In the right context, multi-target modulation is desired or perhaps mandatory for successful therapies [4].
What exactly does polypharmacology mean? Strictly speaking, polypharmacology refers to molecules which are recognized by different molecular targets. The affinity shown towards the targets may vary, but as previously mentioned, such compounds may be discarded fearing this promiscuity may trigger off-target effects [5]. Thus, we are walking a fine line between positive and negative connotations. For that matter polypharmacology usually associates with positive outcomes. It involves the search of “master key compounds” to tackle chronic diseases, for example central nervous system (CNS) disorders share multifactorial processes that ultimately lead to degeneration, physiologically speaking. Therefore, a single target-inhibition is of no use here as complex processes require integral approaches [6].
Compound promiscuity is a concept closely related to polypharmacology. This of course tells us about a molecule that interacts with many proteins or receptors. Promiscuity is usually related to negative connotations, e.g., it is conceptualized as unwanted characteristic such as toxic effects due to off-target interactions. In turn, compound promiscuity is related to the Pan Assay Interference compounds or PAINS. These molecules appear to be a jack-of-all-trades with potent binding and activity, while the truth is they are a master of none. Baell and Waters first warned about these “con artists” as they lure naïve chemists or biologists who waste valuable resources with a lost cause [7]. Of note, PAINS compounds are not always promiscuous. They can be flagged as active because they produce metal chelation, chemical aggregation, redox activity, compound fluorescence, cysteine oxidation or other kind of interference. Putting briefly these concepts together, the pressing matter here is to understand and give the right context to “polypharmacology” meaning that while related to “chemical promiscuity” we cannot put them on the same basket any longer.
Polypharmacy is one more concept related to polypharmacology. Polypharmacy “can mean the prescribing of either many drugs (appropriately) or too many drugs (inappropriately). The term is usually used in the second of these senses, and pejoratively. However, when talking about polypharmacy it would be wise to qualify it as appropriate or inappropriate” [8].
As of 2014 the number of articles citing “polypharmacology” as part of its title and/or as a keyword has increased significantly, with almost 200 articles published in the past three years only [9]. So a multi-target approach is gaining adepts at steady pace. While this shows more promise in the grand scheme of drug discovery we must be careful and correctly asses the opportunities and challenges of this transition era. We should not instantly accept polypharmacology as a panacea of sorts, only time and advances in current knowledge will determine the success of such paradigm change, we must conserve an objective view on the subject with realistic expectations.
Although the road ahead in polypharmacology drug discovery may seem blurry or difficult to achieve, the development of polydrugs is currently possible. As discussed in this review, the development and application of computational methods and tools for in silico drug discovery should be a starting point and compass to navigate the “chemical wilderness”. Computational approaches include, but are not limited to, chemoinformatics, molecular similarity, docking, molecular dynamics, virtual screening and QSAR.
This review is organized in six sections. After this Introduction, general aspects of multi-target vs. target-specific drugs are discussed including the rationale, the ‘master key compound’ concept and safety panels to address the possible unwanted effects of drugs multi-targeting. The next part elaborates on the relationship between polypharmacology and other major concepts in drug discovery, including drug repurposing, combination of drugs and in vivo testing. The section after that describes briefly examples of applications of polypharmacology and polypharmacy to the development of epi-drugs and antiviral compounds, respectively. It follows a discussion of different modern approaches to study systematically polypharmacological relationships and design multi-target drugs. A special emphasis is made on the concept of chemogenomics. The last part of the paper presents conclusions.
2. Multi-target vs. target-specific drugs
As discussed before, the increasing awareness of the large complexity of systems biology is shifting the paradigm in drug discovery from a single-target to a multi-target approach [10]. Despite the fact that the latter approach is significantly more complicated than the one-drug - one-target strategy (largely influenced by a reductionist perspective of systems biology) [11], it may lead to drugs that are more effective in the clinic. However, it has to be considered that multi-target drug design, and polypharmacology in general, highly depend on the dose to deliver an overall clinical benefit [12]. For instance, a drug may have a positive effect at therapeutic doses because of the interaction with multiple targets. However, the interaction of the same compound with anti-targets at higher doses will lead to undesirable side effects [12]. Thus, similar to the appropriate or inappropriate polypharmacy discussed by Aronson [8], polypharmacology can also lead to desirable or undesirable (e.g., unwanted promiscuity) multi-target drug interactions that will depend not only on the nature of the structures of the drugs and targets but also on the compound concentrations. The ‘dual-face’ of multi-target drugs is schematically illustrated in Figure 1.

2.1. ‘Master key compounds’
A ‘master key compound’ (luckily ‘master key drug’) is a molecule that binds to a given number of targets that produce a desirable clinical effect without hitting (or with a minimum effect) off-targets that are related to undesirable, secondary effects [10]. In a simple analogy with a master key, a ‘master key molecule’ should have the ability to operate on a group/set of selected targets (doors) but not on any ‘doors’, in particular those anti-targets that lead to undesirable side effects. Table 1 illustrates examples of master key drugs that are used in the market. The table summarizes the name, chemical structure, clinical use and the associated molecular target receptors.

Kinase inhibitors are representative yet controversial examples of master key compounds used in the clinic. Despite the fact there are differences in the kinase domains, the binding site of ATP is highly conserved across all the kinases. Since the ATP site is targeted by a large number of kinase inhibitors, there are selectivity issues and there is a significant challenge to develop master key inhibitors of kinases (Figure 2). Several efforts in the pharmaceutical industry and academia have been dedicated to develop selective kinase inhibitors in order to reduce side effects. However, it is also noteworthy that some extremely promiscuous kinase inhibitors have shown good clinical performance, even when treating unrelated tumors, for instance dasatinib which binds to at least 159 kinases [13]. This has not been overlooked by the scientific community, which is already working on the idea of designing multitarget protein kinase inhibitors [14,15]. Currently, there are two strategies to exploit polypharmacology against kinases, i.e. combination of selective compounds and design of ‘selectively nonselective’ i.e., master key kinase inhibitors (Figure 3) [10,14]. The former refers to the simultaneous administration of two selective compounds designed to inhibit different kinases in order to achieve an enhanced phenotypic effect. The latter, and the most difficult, consists in merging the inhibitory activity against two or more kinases in one single compound with none (or only few) off-targets. The key factor in either of the mentioned strategies is to identify the targets that should be inhibited simultaneously to produce a selective phenotypic effect against the tumor.
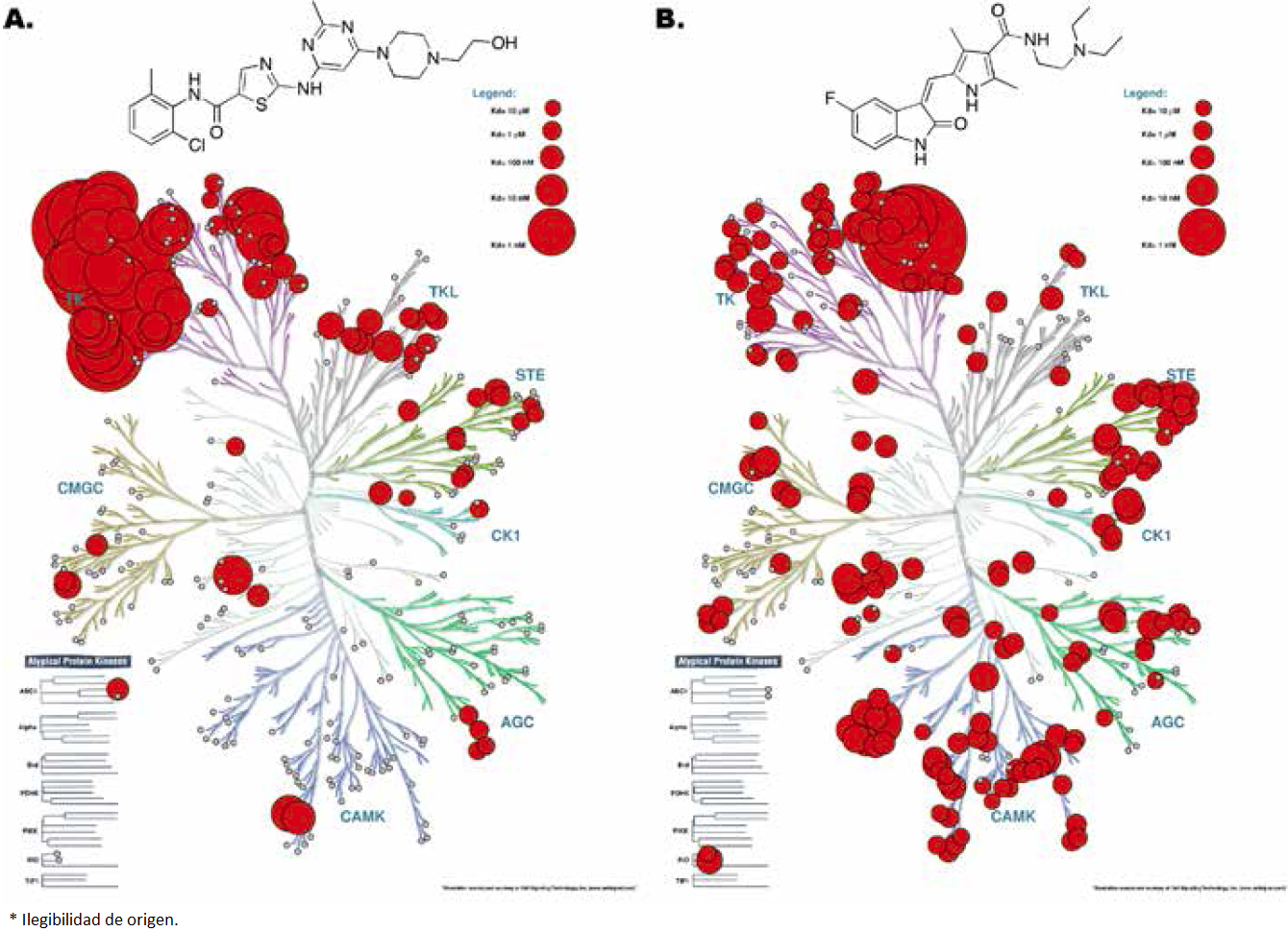
Figure 2 Examples of poor selectivity of kinase inhibitors. This figure shows the cross-reactivity of dasatinib (A) and sunitinib (B) across the kinome. Data was obtained from Karaman et al. [16] and the figure was generated using Kinome Render [17].
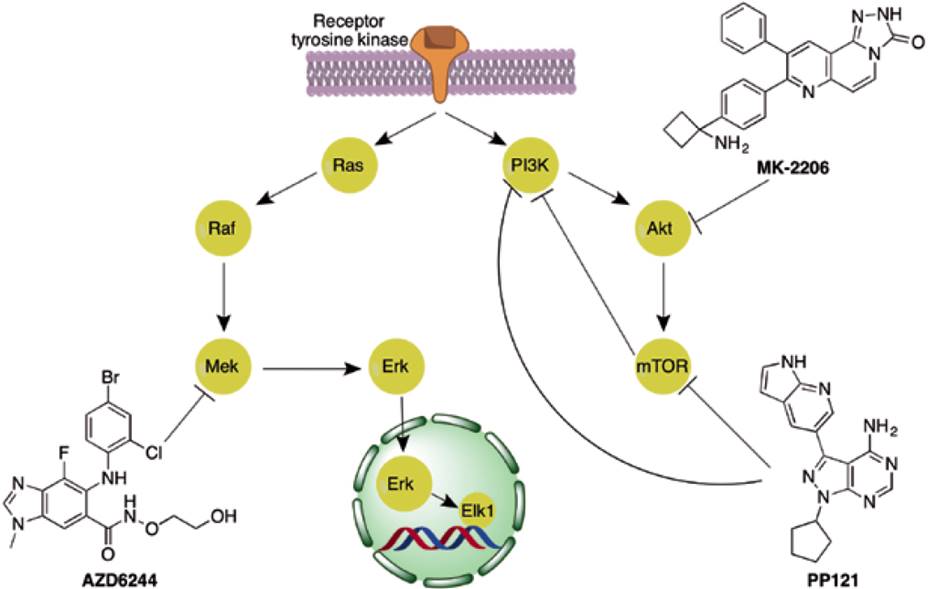
Figure 3 Examples of the use of polypharmacology against kinases. Compounds AZD6244 and MK-2206 have been used in combination to inhibit the MAPK and PI3K pathways to obtain an enhanced phenotypic effect. Compound PP121 inhibits both PI3K and mTOR simultaneously. This dual inhibition has been proposed to be more potent than inhibiting either target individually. The rationale behind this idea is that mTOR activates a negative feedback loop that inhibits PI3K. The inhibition of mTOR alone results in the blockage of the negative feedback loop and in a hyperactivation of PI3K [18].
2.2. Safety panels
Many of the adverse drug reactions (ADRs) are caused by unintentional interaction of a drug with a non-therapeutic target to which is given the name “antitarget”. The most frequently found antitargets are already well studied and characterized. Examples of these receptors are shown in Table 2.
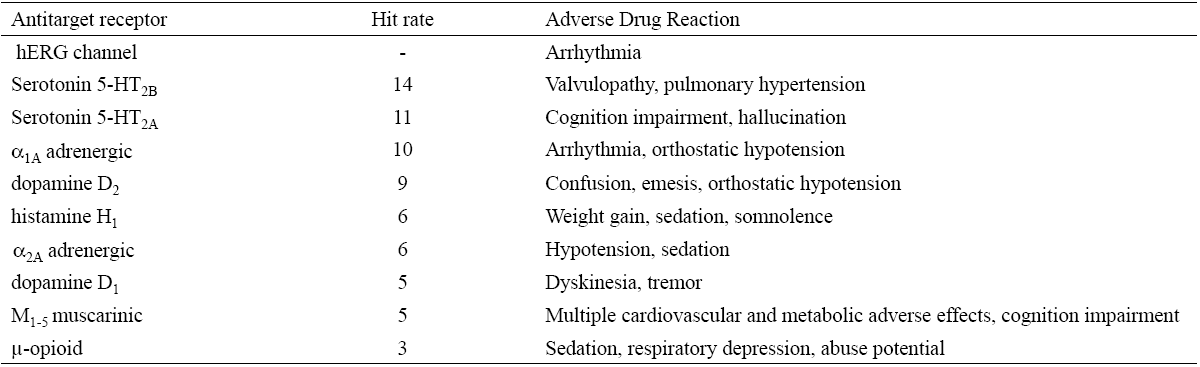
Animal toxicity models are not practical to predict the adverse effects caused by antitargets in humans due to differences between species. For instance, human ion channels differs greatly from their rodent orthologues. Therefore, the ICH (International Conference on Harmonization) guidelines S7A for Security Studies, recommend performing antitarget screening tests. These tests are ligand binding assays using enzymatic methods to obtain data and provide suggestions concerning potential adverse effects of the molecule under study. Antitarget screening tests also protects the early volunteers in clinical studies from developing ADRs as it is estimated that the toxicity in the preclinical stage causes that 30% of the compounds do not advance to the clinical stage.
3. Polypharmacology and related concepts in drug discovery
The interaction of a compound with multiple targets is at the core of several major concepts in current drug discovery [10]. Herein we further elaborate on these relationships.
3.1. Drug repurposing
In general terms, drug repurposing or repositioning is the identification of a new clinical use for compounds that have already proven to be useful to treat a defined medical condition. It can extend only to the conceptual approach or involve the whole process, until the new indication is approved by the respective institutions. Notably, drug repurposing is not by itself a strategy, since it may be a result of different strategies and methods; it can be serendipitous, semi-systematic, or fully systematic, by means of high-throughput screening or in silico approaches [19]. The subjacent principles of drug repurposing imply that drugs might have activity against more than one therapeutic target, i.e., polypharmacology.
Nonetheless, there are many distinct definitions of drug repurposing, and there are efforts focused on further condensing the different proposals. Recently, Langedijk et al. performed a systematic review of the literature in order to unify the otherwise diverse and sometimes discordant definitions of “drug repurposing / repositioning / redirecting / reprofiling / rediscovery” and concluded that the main features of these definitions are regarding [20]:
a) General concept: strategy, process, approach;
b) Action performed: identifying, using or developing;
c) Innovative use: for a different disease, patient population, dosage or route of administration;
d) The product itself: an existing or abandoned pharmaceutical active ingredient, patent, medicinal product, and so on.
In particular, systematic or rational drug repurposing is of current general interest, due to the marked advantages of drug repurposing versus conventional drug discovery in terms of time, costs, and patients’ safety [21]. More specifically, computational approaches have proven to be cost-effective, and are viable options under several circumstances, such as finding therapeutic agents against neglected [22-24] or rare diseases [25]. In several recent studies potential alternative activities are being uncovered and further investigation is underway to see if these compounds can be approved for clinical use for the alternative indication. e.g., olsalazine, a drug approved for the treatment of inflammatory bowel disease, was recently identified as a novel hypomethylating agent using a chemoinformatic-based virtual screening approach [26]. Concisely, olsalazine is being investigated as a potential epigenetic drug. Following up this successful proof-of-concept, additional computational studies have been conducted with the aim of repurpose approved drugs as potential epi drugs [19].
3.2. Combination of drugs
Combinations of drugs are clinically relevant for treating a variety of chronic medical conditions, such as infectious, metabolic, malignant or neurological diseases [8]. A clear example is the Highly Active Antiretroviral Therapy or HAART used for the treatment of patients infected with the human immunodeficiency virus HIV [27]. Combinations of drugs could be used to prevent or attack resistance to single agents and to improve the clinical effect of the treatment. However, this approach often ends with polypharmacy (the intake of five or more drugs), a well-described clinical condition that can lead to increased risks and adverse effects from medications, especially in the elderly or patients with multiple chronic diseases [28, 29].
In the scientific literature, it is generally conceived that the development of polypharmacological agents is the next logical step once it is known that a single chemical compound may affect multiple biological targets (e.g., adverse or off-target effects) and that combinations of drugs that act on different targets might have additive or synergic effects against a disease. Polypharmacology is believed to be a promising feature of drugs that could replace combined drug therapies [30] and thus avoid polypharmacy.
Nonetheless, there is another point of view in which drug combinations are included within polypharmacology approaches following the multi-target paradigm, although still recognizing the advantages of multi-target single agents [31]. A third approach was developed recently by Gujral et al. after this group identified kinases involved in cellular migration that are specific for cell type. To accomplish this, they tested polypharmacological kinase inhibitors. Their proposal is to exploit polypharmacology of chemical probes to aid in the rational design of more potent and specific drug combinations [32]. This last approach is supported by the finding that combination therapies acting synergistically are also more specific in their pharmacological actions when administrated in combination than as single agents [33]. Hence, combination of drugs and polypharmacology does not necessarily imply more severe adverse effects when there is a synergic effect, provided that selectivity is increased in these cases.
Finally, the combination of drugs may be used for preventing adverse effects or severe risks of certain drugs used in monotherapy. For example, Zhao et al. discovered that co-administration of exenatide substantially reduces the myocardial infarction risk found in diabetic patients treated with rosiglitazone alone. Both exenatide and rosiglitazone are indicated for the treatment of diabetes mellitus type II and they act on different targets [34].
3.3. In vivo testing
Several drugs have been identified following an in vivo screening or natural product mixtures or mixtures of individual compounds. In vivo testing is a drug discovery approach that distance itself from the ‘classical’ one-target-screening. It has been recognized that in vivo screening offers the advantage of an early demonstration that compounds may show activity in disease-relevant models before proceeding with further development. Moreover, despite the limitations and costs of in vivo testing, it allows the rapid selection of molecules that exert their biological effect through the interaction with multiple-targets (present in an in vivo system). Therefore, this methodology represents an approach to identify ‘master key compounds’ discussed above. In vivo testing of mixture-based combinatorial libraries has been used as an effective drug discovery approach to rapidly screen hundreds or thousands of compounds efficiently [35]. Moreover, in vivo testing of mixture-based combinatorial libraries has enabled to expand the exploration of the chemical space beyond the one populated by currently marketed drugs [10].
4. Polypharmacology (and polypharmacy): case studies
In this section we discuss briefly selected applications of polypharmacology and polypharmacy for the treatment of diseases associated with epigenetic alterations and antiviral infections caused by the human immunodeficiency virus (HIV), respectively. Both types of diseases are different in nature but both are very complex and represent major challenges to design effective therapeutic treatments.
4.1. Polypharmacology in epigenetics
For diseases with a complex metabolic substrate, such as diabetes, cancer, autoimmune and neurodegenerative disorders, it becomes increasingly evident that aiming at a single pharmacological target would not be an appropriate strategy. It has been shown to a variable extent for each of the aforementioned diseases, that epigenetics (inheritable traits that are not encoded within the genome; e.g., DNA methylation and histone modifications) plays an important role in the establishment and maintenance of the disease [36]. Epigenetics mechanisms are extremely complex, and not yet totally understood, which makes quite difficult to design therapies directed against them. However, epigenetic drugs are appearing in the clinical scenario, mostly for the treatment of malignant or pre-malignant states, with favorable results [37, 38]. Moreover, there is a current trend for shifting towards epi-polypharmacology drugs, against either more than one epigenetic target or combined epigenetic and other targets [4]. Notably, many different epigenetic biological targets share a reduced number of cofactors (e.g., Zn+2, NAD+, SAM), and thus it is feasible to guide the design of cofactor inhibitors with polypharmacologic properties [39]. Another approach is the design of hybrid molecules. This strategy has led to the development of pan-demethylase inhibitors by synthesis of hybrid molecules containing inhibitors of histone demethylases LSD1 and JmjC, thus generating compounds that increase H3K4 and H3K9 methylation levels and produce apoptosis selectively to cancer cell lines, with little effect on non-cancer cells [40]. In other cases, compounds that are likely to inhibit concise epigenetic targets show polypharmacology against other epigenetic targets. This was the case of the AMI-5 analogues synthesized by Mai et al [41]. AMI-5 was described previously as a small molecule inhibitor of protein arginine and histone lysine methyltransferases, whereas some of its analogues were able to target multiple epigenetic targets, including protein and histone methyl and acetyltransferases.
4.2. Charting the epigenetic relevant chemical space
As discussed in the preceding section, epigenetics involves a series of complex phenomena involving different enzymes that work as readers, erasers and writers. Towards the design of compounds directed to multiple epigenetic targets, we have initiated a first assessment of the epigenetic relevant chemical space (ERCS) focused on DNA methyltransferase inhibitors [42]. To further illustrate this point Figure 4 shows a visual representation of the chemical space obtained by principal component analysis (PCA) of six physicochemical properties of data sets of molecules tested as inhibitors of bromodomains, histone deacetylases, and DNA methyltransferases. As reference, Generally Recognized as Safe (GRAS) molecules were included. The physicochemical properties computed were number of acceptors/donors of hydrogen bonds, number of rotatable bonds, molecular weight, octanol/water partition coefficient and topological surface area. According to this visualization, different compound data sets populate similar regions in the chemical space since they share similar physicochemical characteristics. From this preliminary analysis it can be expected that ERCS have a substantial overlap with the chemical space of other small molecules used in drug discovery of the food industry.
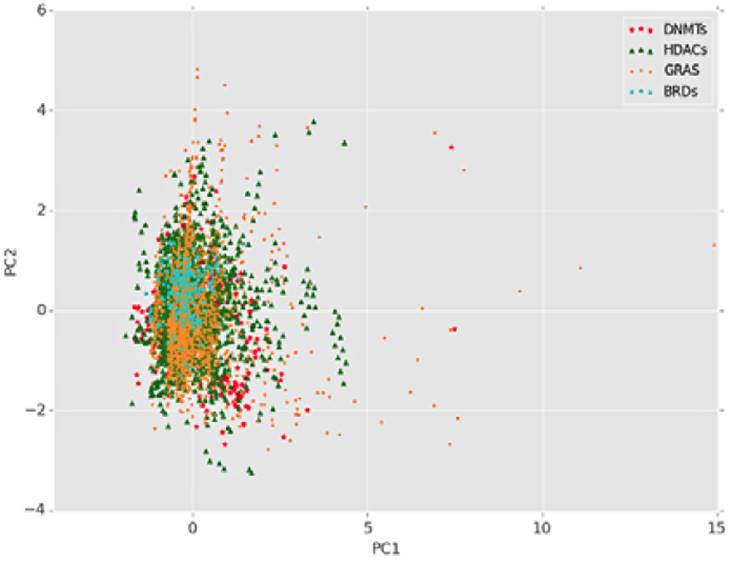
Figure 4 Visual representation of the chemical space of inhibitors of histone deacetylases (HDACs), bromodomains (BRDs), DNA methyltransferases (DNMTs), and Generally Recognized as Safe (GRAS) compounds. The principal component analysis was done with six pharmaceutically relevant physicochemical properties. The first two principal components (PC) are represented in the figure.
It is expected that the mapping and interpretation of chemical space improves the current knowledge on epigenetics. For example, by assessing the properties of epi-compounds it may be possible to develop empirical rules to catalog new molecules as epi-modulators and, overall, identify the structural characteristics needed to achieve optimal multiple-epi-inhibition [43].
4.3. Polypharmacy for the treatment of HIV-infections
Acquired Immune Deficiency Syndrome (AIDS) is still a major health problem. In 2014 there were 36.9 million people living with the human immunodeficiency virus (HIV). The drugs available today for the treatment of HIV can be classified in several classes: reverse transcriptase inhibitors: nucleoside (nucleotide) (NRTIs) and non-nucleoside (NNRTIs); HIV protease inhibitors, integrase inhibitors, a fusion inhibitor (to prevent the fusion of the viral envelope with the host-cell membrane), and a CCR5 inhibitor (to block the interaction of the virus with one of its receptors at the host cell) [44]. The rapid emergence of resistant strains requires the co-administration of several drugs with different mechanisms of action and hitting different molecular targets. HIV-infected individuals are subject to the Highly Active Antiretroviral Therapy (HAART) where two or more drugs are administered in various combinations and administration schedules. Current treatments require the combination of at least two or three active drugs from at least two different classes. Despite the fact that this polypharmacy approach is able to reduce the viral loads in patients, reducing the incidence of opportunistic infections and deaths in AIDS patients, there are concerns of serious side effects and the eventual fail of a given treatment schedule due to the emergence of resistance. Resistance is primarily due to the development of mutations in RT, integrase, and HIV protease. Moreover, Edelman et al. discusses that indeed polypharmacy is the next therapeutic challenge in HIV [29].
5. Computational strategies to explore polypharmacology
Since the chemical and biological spaces are huge, the relationship between the two spaces is highly complex. Therefore, in order to describe, understand, and ideally predict the relationship between the two spaces, efficient computer-based methods are necessary. In response to such need chemogenomics has emerged as a multidisciplinary research field. A number of rich reviews have been published on chemogenomics [45, 46]. In this section, we discuss recent developments on chemogenomics followed by representative and specific computational chemogenomics methods.
5.1. Chemogenomics: intersection of chemical and biological spaces
The concept of polypharmacology is at the interface of the chemical and biological spaces. Both the chemical and biological spaces are intuitive concepts because of its analogy with the cosmic universe [47, 48]. There are several definitions of chemical space. For instance, Virshup et al. define chemical space as ‘an M-dimensional Cartesian space in which compounds are located by a set of M physicochemical and/or chemoinformatic descriptors’ [49]. The concept of chemical space has a broad application in drug discovery that can be classified in two major groups: 1) classification of bioactive compounds depending on their therapeutic target or associated pharmaceutical effect, and 2) compound library design and selection. By analogy, biological space can be understood as the set of all possible targets. Some of them, however, are associated with a desirable chemical effect, others are related to off-targets leading to adverse effects, “orphan targets” (for which no compounds/drug have been identified yet), and targets to be identified.
Chemogenomics is a multidisciplinary research field that aims to identify the possible associations of all possible ligands for all possible targets [50]. To achieve this goal a number of in vitro and in in silico approaches are employed [45]. In other words, chemogenomics aims to find the association between the chemical and target spaces or to characterize the intersection between chemical and biological spaces. The concept of chemogenomics is schematically illustrated in Figure 5. As discussed in detail elsewhere, chemogenomics is highly associated with concepts such as polypharmacology itself, drug repurposing, in vivo high-throughput screening, pharmaceutical profiling, virtual screening, target fishing, and structure-multiple activity relationships (SmAR), (see below).

Figure 5 Schematic representation of a chemogenomics matrix; the rows represent all possible compounds and the columns represent all possible molecular targets.
Chemogenomics data sets are major resources to conduct systematic studies to find associations between compound-target interactions. Table 3 summarizes examples of chemogenomics data sets [51-60]. One of the current limitations of these data sets is that they are still rather incomplete. For instance, in order to analyze drug-target interaction networks it has been analyzed the effect of the lack of data completeness which has been called the “Achilles Heel” of drug-target networks [61]. However, such databases are rich sources of information to describe ligand-target interactions and to uncover new target-ligand relationships. Other major area of improvement of chemogenomics data sets are the so-called ‘five I’s’: data may be incomplete, inaccurate, imprecise, incompatible, and/or irreproducible as recently described by Fourches et al. [46].
Authors of that work proposed a general workflow to conduct chemical and biological data curation [46]. Figure 6 illustrates in a schematic manner examples of computational approaches employed to explore chemogenomics relationships [62-64].

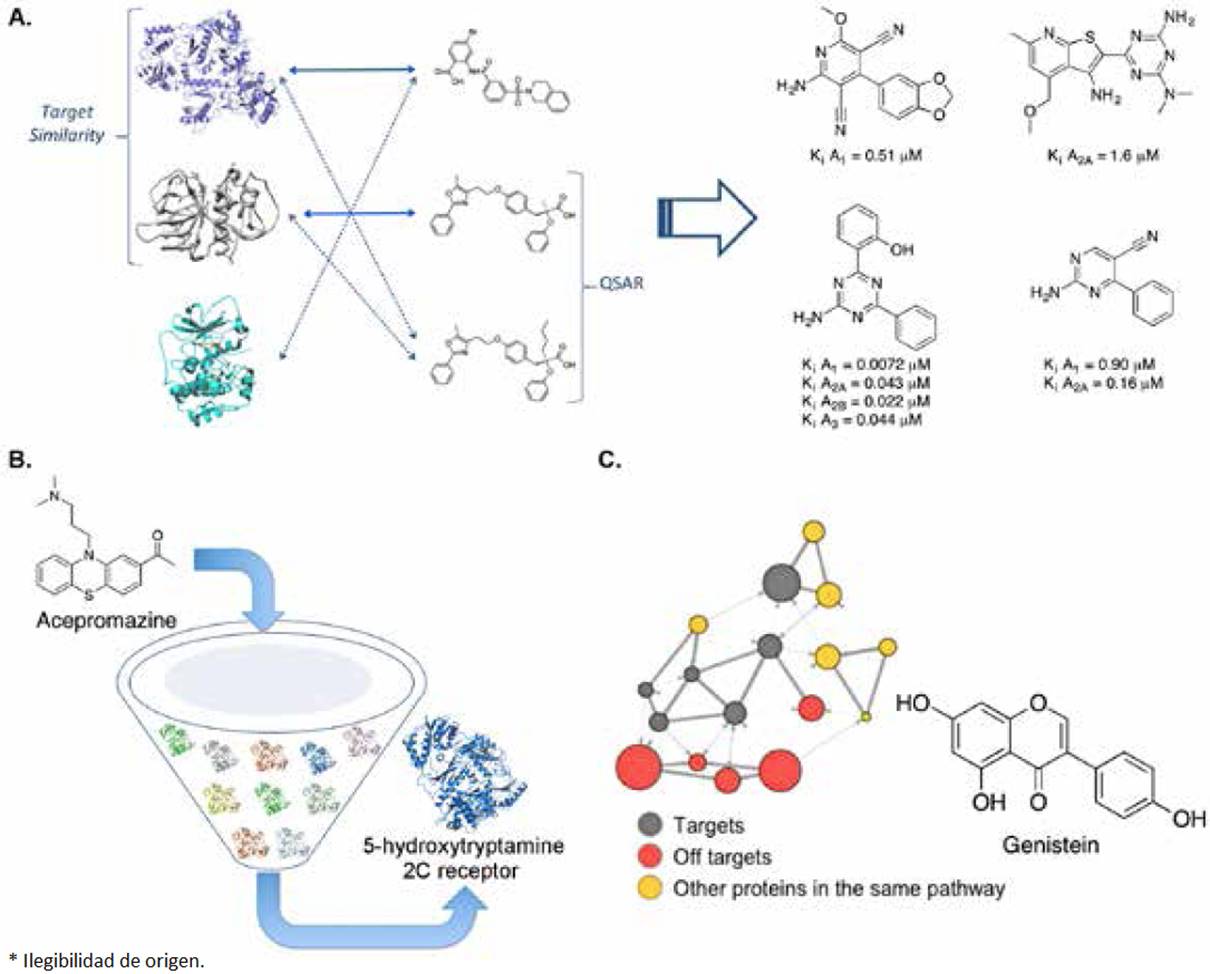
Figure 6 Schematic representation of some computational strategies used to explore chemogenomics. Panel A shows an example of proteochemometric model, which combines target and structural similarities to predict activity of new compounds against adenosine receptors [62]. Panel B represents target fishing, which is commonly used to identify targets of known compounds such as in the case of acepromazine [63]. Panel C represents an example of systems pharmacology that was previously used to identify the anti-cancer activity of genistein [64].
5.2. Structure multiple-activity relationships
Drug discovery based on one molecule one-target approach gave rise to biological assays where, typically, one compound is associated with one measure of activity. In order to establish the corresponding structure-activity relationships, several methods have been developed including qualitative and quantitative approaches. Outstanding examples are the quantitative structure-activity relationships - QSAR [65]. A second, more recent approach is the activity landscape modeling (ALM) aimed to identify the relationship between structure similarity (given a set of molecular representation) and activity similarity [66, 67]. Over the last few years, QSAR, ALM and other computational approaches are being adapted and developed to identify and predict Structure-multiple Activity Relationships (SmARs) that emerge when compound data sets are screened across a range of molecular targets [68, 69]. For instance, our research group has reported the SmAR of benchmark data sets screened across multiple targets of therapeutic interests and bioassay data obtained from PubChem [68, 69]. In both case studies, structure-activity similarity (SAS) maps, that systematically relate structure similarity with potency difference for each pair of compounds, were adapted to represent multiple activity-similarities [67]. Thus, the idea of measuring activity similarity can be applied not only to SAS maps but to basically any other activity landscape model.
5.3. Proteochemometric modeling
Proteochemometric (PCM) modeling can be conceptualized as an extension of QSAR modeling that exploits chemogenomic data by performing a quantitative evaluation of ligand and target structural similarities. As a result, this technique allows the simultaneous navigation, inter- and extrapolation in both chemical space (i.e. ligands) and biological space (i.e. protein target) [70, 71]. By the explicit combination of target and ligand information in a single model, PCM is capable to analyze and predict SmARs of a set of compounds [70, 71]. It has been shown that PCM is better suited for the prediction of SmARs than other methods such as fragment-based models [72] and multitarget-QSAR using support vector machines (SVM) [73]. This technique has been successfully applied to study the SmARs of different target families such as GPCRs [62], cytochrome P450 isoforms [74], serine proteases [75], among others [76-79]. This method is also well suited for the study of kinase selectivity profiles and has been applied to a large number of data sets [72, 80-83]. In general, predictive models that include ligand and target information represent a step forward for the analysis of multitarget inhibitors as they usually achieve better performance compared to single target methods. All details of PCM modeling requirements and more applications have been reviewed recently [70,71].
5.4. Target fishing
The goal of the approach commonly called as ‘target fishing” is to uncover biomacromolecules or molecular targets that are able to bind to a given ligand (or drug). Many techniques, including computational approaches, can be adapted to carry out the inverse process of traditional virtual screening (Figure 5). In other words, the biological target space is interrogated to identify potential targets for (typically) a small molecule.
A typical computational technique employed in target fishing is molecular docking giving rise to the ‘inverse docking’ strategy [84]. In this technique, introduced by Chen and Zhi [85] a given small molecule is docked across a data base of 3D macromolecular targets. An alternative computational approach used in target fishing is data mining. Other approach is to measure the molecular similarity between the compound of interest and a data set of known ligands of molecular targets (for instance, the structure of co-crystal ligands). These methods and other approaches have been recently reviewed in a rich paper by Cereto-Massagué et al. [86]. Of note, this review includes comprehensive lists of molecular databases and web resources useful for in silico target fishing.
As in traditional virtual screening, the chemical compounds can have basically any origin such as novel chemical synthesis, commercial libraries or natural products, to name few examples. Natural products are, perhaps, one of the most studied molecules using this approach [87]. Importantly, in the context of polypharmacology, approved drugs or compounds in clinical trials can also be subjects of target fishing. In fact, prediction of molecular targets has become an active area or research in drug repurposing [86]. Target fishing of approved drugs can have one or more goals depending on the specific study:
a) To identify the molecular targets of a drug for which is uncertain the mechanism of action.
b) For those drugs with known action mechanism to identify additional molecular targets that produces a beneficial clinical effect. In other words, to explore in a systematic manner polypharmacology.
c) Uncover off-targets in a systematic manner. This can lead to the prediction of secondary effects.
Examples of the application of target fishing using natural products as query compounds have been reviewed by Medina-Franco [87]. Several recent examples towards target fishing for drug repurposing have also been published [88-90].
5.5. Data mining of side effects interactions for drug repurposing
Drug repurposing through data mining has two principal premises: 1) there is vast information (e.g., clinical, phenotypical, and experimental) regarding the drugs that are intended to be repurposed; and 2) the obtained information is sufficient to fit a statistical model to predict whether a compound would be active against another target or disease. Text similarity is a particularly developed tool in these settings.
Through text similarity searching, the scientific literature can be mined, in order to link, often indirectly, drugs and diseases by association of terms [91]. A clear example is data mining of adverse effects; this approach assumes that drugs with similar adverse (also called off-target) effects may be active against similar diseases. Within this pipeline, Campillos et al. developed a model for drug repurposing with efficiency rates higher than 50% [92]. Notably, comprehensive online databases have been developed to address the problem of disperse information about drugs; these resources are a result of scientific literature mining and contain complete references about different compounds [93, 94].
5.6. Systems pharmacology
Systems pharmacology has arisen as an emerging trend strongly connected to polypharmacology. The main similarity between these two lies in their fundamentals: both try to overcome the simplicity of the old-fashioned “one drug, one target” paradigm. Polypharmacology has often the connotation of “one drug, more than one target”, implying both the possibility of drug repurposing and the feasibility of multi-target treatments with a sole drug, as we have explored throughout this manuscript. However, systems pharmacology is a wider concept than polypharmacology, described by the phrase “one treatment, one network”. Thereby, the focus of systems pharmacology implies the rational design of therapies accounting for the overall cellular and physiological complexity, aiming to biological networks rather than isolated targets [95]. The two main strategies emerging from systems pharmacology are 1) those based on simulations in interaction networks validated in the scientific literature [96, 97] and 2) approaches exploiting high-throughput data such as expression or genetic micro-arrays [98-100]. Both approaches aim to find a differential function of pathways in pathologic processes compared to healthy states, and drugs that can reverse the pathogenic features. Therefore, the objective is to develop treatments that avoid the studied pathological phenotypes [97,100].
5.7. Polypharmacology fingerprints
As part of the computational strategies to explore and eventually predict polypharmacology, Pérez-Nueno et al. have developed a computational polypharmacology fingerprint based on the Gaussian ensemble screening approach developed before by the same authors [99]. The newly developed fingerprint was designed to encode information related to promiscuity. In that work, the fingerprint was built using about 800 established drug targets from a public database of known drugs. In a benchmark study, the proposed fingerprint was able to predict up to 90% of the experimentally known polypharmacology associations (with no missing data). Finally, in the work the authors demonstrated that the proposed fingerprints represent a new approach to suggest molecular targets for preclinical compounds and clinical drug candidates. As the authors described in the excellent paper, the polypharmacology fingerprint represent an important addition to other in silico tools based on different type of descriptors that are intended to relate quantitatively biomolecular targets (e.g., protein receptors) to each other (either by computing similarity between the ligands or between the targets) [101].
6. Conclusions
To better understand and potentially predict polypharmacology, it is necessary to explore the intersection between the chemical and biological spaces. One approach to explore such intersection is through the emerging research field of chemogenomics. To date, there are chemogenomics data sets available to conduct drug repurposing, several in the public domain. A major challenge while working with these chemogenomic resources is that the data may be incomplete. Also, it has been recently emphasized the need to conduct curation of the chemical and biological information. A broad range of novel computational strategies are being developed and implemented to mine, understand, and predict polypharmacology. For instance, proteochemometric modeling and multi-target activity landscapes enable the simultaneous analysis of chemical and biological relationships. Using structure or ligand-based approaches, target fishing aims to identify potential targets for a given ligand. Data mining of side effects and systems pharmacology are further examples of novel approaches employed in polypharmacology. Polypharmacology is a promising avenue in emerging and complex drug discovery strategy such as the development of epi-drugs.

