











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Polybutadiene (PB) has been an important focus of research effort during the last half-century due to its vast field of applications arising from its low temperature resistance, as well as the allylic hydrogen atom and the weakly double bonds which provide the possibility of grafting and crosslinking, in addition to its easy tunable properties which can significantly vary according to the micro- and macrostructure. According to the employed catalyst system different types of PB can be obtained; being high cis-PB (≥92% cis-1,4 content) produced by means of coordination polymerization using different metal catalysts such as titanium, cobalt, nickel and Neodymium (Nd) [1-4], each of which generate polymers that vary considerably in terms of macrostructure, which is mainly dependent on the metal catalyst’s ability to undergo chain-transfer reactions. In this matter, early transition metals such as titanium based catalysts are less likely to participate in beta-hydride chain transfer reactions than the more electron-rich late transition metals, allowing the formation of polymers with narrow molar mass distributions index and branching degree. Late transition metals on the other hand such as nickel and cobalt systems are dominated by metal hydride chain transfer reactions, thus leading to polybutadienes with very large molar mass distributions, and high branching degree. As far of the Neodymium systems, their macrostructure is mainly controlled by the propensity of the catalyst to chain transfer reaction to the aluminum co-catalyst [5], distinguishing the formed polybutadienes for being linear polymers but with broad molar mass distributions, giving them, together with their very high cis content of around 98%, very special properties such as excellent abrasion and fatigue resistance besides easy processing, which are taken as advantages for specific applications such as in the tire industry where they are blended with polyisoprene and/or SBR and applied in either sidewalls, threads or rims of tires [6].
There has been reported two types of Nd catalyst systems: ternary systems comprising a Nd carboxylate, an alkylaluminum and a halogen source, or binary systems constituted by Neodymium halide complexed with three equivalents of a Lewis base such as alcohols or any other electro-donor organic compound and a trilalkylaluminium as co-catalyst [7,8]; the behavior of both types of catalyst systems is rather similar, however are the ternary systems which have been more extensively studied, leaving a huge field of research concerning the binary catalyst systems to be developed. Contributing to this field, Zhang et al., 2007 [9] reported the binary catalyst system NdCl3·3EHOH (2-ethylhexanol)/TEA for isoprene polymerization; finding a higher catalytic activity than the ternary system NdV/DEAC/DIBAH and a cis-1,4 structure content of around 96% which could be easily tuned by playing with the Al/Nd molar ratio. Masuda et al., 2002 [10] on the other hand employed a catalyst system comprised by Nd isopropoxide (Nd(Oi-Pr)3 activated by MAO, yielding a high efficient polymerization with a moderate high cis-1,4 stereospecificity (around 90%), which increased to up to 95% by using chlorinated co-catalysts such as DEAC. Rao et al., in 1999 [11] introduced the catalyst system Nd tripentanolate (NdCl3⋅3L, where L = 1-, 2- or 3-pentanol) using TEA as co-catalyst, which allowed the generation of polymers with 99% of cis-1,4 structure. Furthermore it was found that this catalyst allowed also a proper tuning of the micro- and macrostructure by varying the ratio of the catalyst and co-catalyst, and lead to catalytic activities higher than even many of the ternary Nd based catalyst systems. Taking this as reference, and considering the significant role of the type of co-catalyst due to the type of termination that Neodymium-based catalysts undertake, our group has intended to investigate further this catalyst system, looking forward to get a better understanding about the behavior of this catalyst system, and find the optimal reaction conditions. Recently we studied also the polymerization of butadiene by means of NdCl3 tripentanolate and TIBA as co-catalyst, focusing on studying the effect of the catalyst aging time and the reaction temperature [12]. In this research work it was evaluated different co-catalysts (TIBA, TEA, DIBAH and MAO) investigating in detail the role of them played over the catalytic activity, micro- and macrostructure, as well as the thermal properties of the resulting polymers.
According to the current knowledge, the proposed mechanism of formation of active sites is comprised by firstly an alkylation reaction followed by the complexation between the Neodymium compound and an organoaluminum, as represented in Fig. 1.
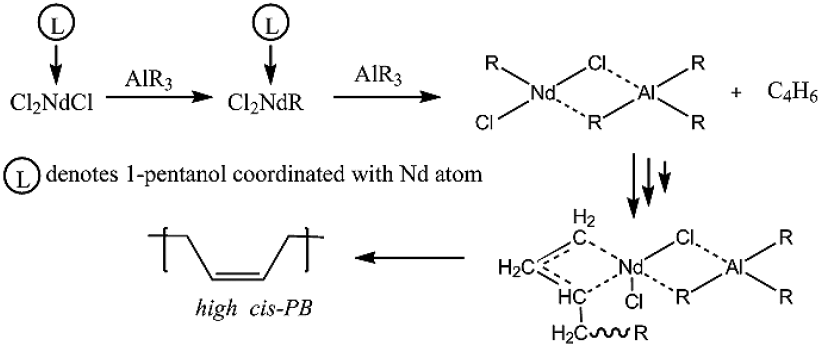
Fig. 1 Mechanism of the active center formation and synthesis of high cis-1,4 PB by means of NdCl3·3L with L = 1-pentanol
In addition, it is noteworthy the variety of products which might be formed in this complexation step, resulting in different active sites which vary not only in structure, but also in kinetic behavior. The proposed structure of the possible active sites reported in the literature is as shown in Fig. 2 [13].
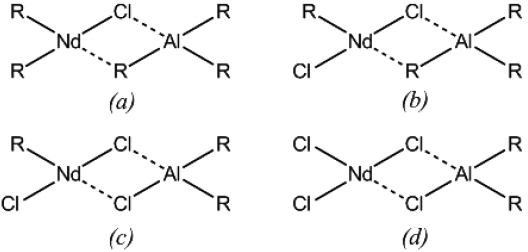
Fig. 2 Catalytic complexes that may are present in the polymerization, (b) and (c) corresponds to active centers according to Manuiko et al. [13]
Results and Discussions
A series of experiments related to the synthesis of polybutadiene catalyzed by neodymium chloride tripentanolate (hereafter called catalyst) and the different co-catalysts in cyclohexane at fixed Al/Nd molar ratio of 35, aging time of 15 minutes at 30°C and a polymerization temperature of 50 °C are shown in Table 1.
Table 1 General properties of the resultant polymers.
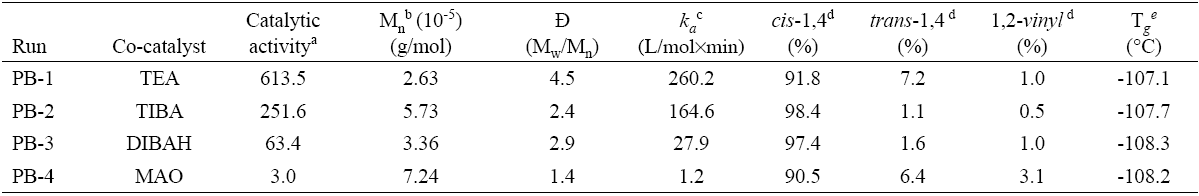
* Note: isothermal polymerizations were performed in cyclohexane at 50°C; [cyclohexane]0 = 8.44 mol/L; [1,3-butadiene]0 = 1.05 mol/L [NdCl3·3L]0 = 0.547 mmol; [Al]/[ Nd] = 35; aging temperature = 30 °C; aging time = 15 min.
a kg PB/mol Nd h.
b Determined by size exclusion chromatography (SEC) with polystyrene standards.
c The apparent first order rate constant k a was calculated considering the kinetic law - d[M]/dt=k a [Nd][M] and from plots ln(1-x)-1 = f ( t ), where x is the monomer conversion.
d The microstructures were determinate by 1H and 13C NMR spectroscopy.
e Determined by differential scanning calorimetry (DSC).
Important differences were found in the polymerization behavior among the co-catalysts, in the way that by far the highest catalytic activity was exhibited by using TEA, with which 80% of monomer conversion was already observed after only 8 minutes; followed by TIBA which after 20 minutes reached 82% of monomer conversion. With DIBAH on the other hand, 62% of monomer conversion was observed after 60 minutes, and lastly MAO generated the lowest catalytic activity and only 3% of monomer conversion after 60 minutes of reaction. These differences between the catalytic activities imply a significant difference in availability of active centers able to initiate the polymerization, which seems to be governed by the different alkylating capacity of the organoaluminum species [14]. The very low catalytic activity observed with MAO as co-catalyst could be attributed to a possible coordination of the alcohol ligand to MAO which led to destabilization of the active site, as it has been reported before for other catalyst systems activated also by MAO [15, 16]. It is also noteworthy that the trend of catalytic activity behaved inversely proportional to the bulkiness of the substituents in the co-catalyst, being TEA which led to the highest catalytic activity, substituted by simple ethyl groups. This implies that the steric hindrance and electron-releasing effect of bigger or more polar substituents destabilize the catalytic complex. The Fig. 3 shows the plots of ln(1-x)-1 vs polymerization time for different co-catalyst and can be seen in all cases a linear relation, indicating that the polymerization rate can be described by first order kinetics with respect to the monomer concentration, which is in agreement to the reported in the literature for butadiene polymerizations using neodymium based catalysts [17-19], Furthermore it is possible to evaluate the apparent first-order rate constant (k a ) and the calculated values are summarized in Table 1.

Regarding the molecular weight characteristics summarized in Table 1, it was shown a decreasing tendency by increasing the catalytic activity, since greater amount of available active sites results in a decreased molecular weight of the polymer so formed. On the other hand the decrease in molecular weight was accompanied by an increase in polydispersity index (Mw/Mn) showing that the termination was governed by transfer reactions, being to the organoaluminum compound the characteristic in Nd based systems, leading to linear polymers but with broad molecular weight distributions. This behavior was found to be intensified significantly by employing TEA as co-catalyst.
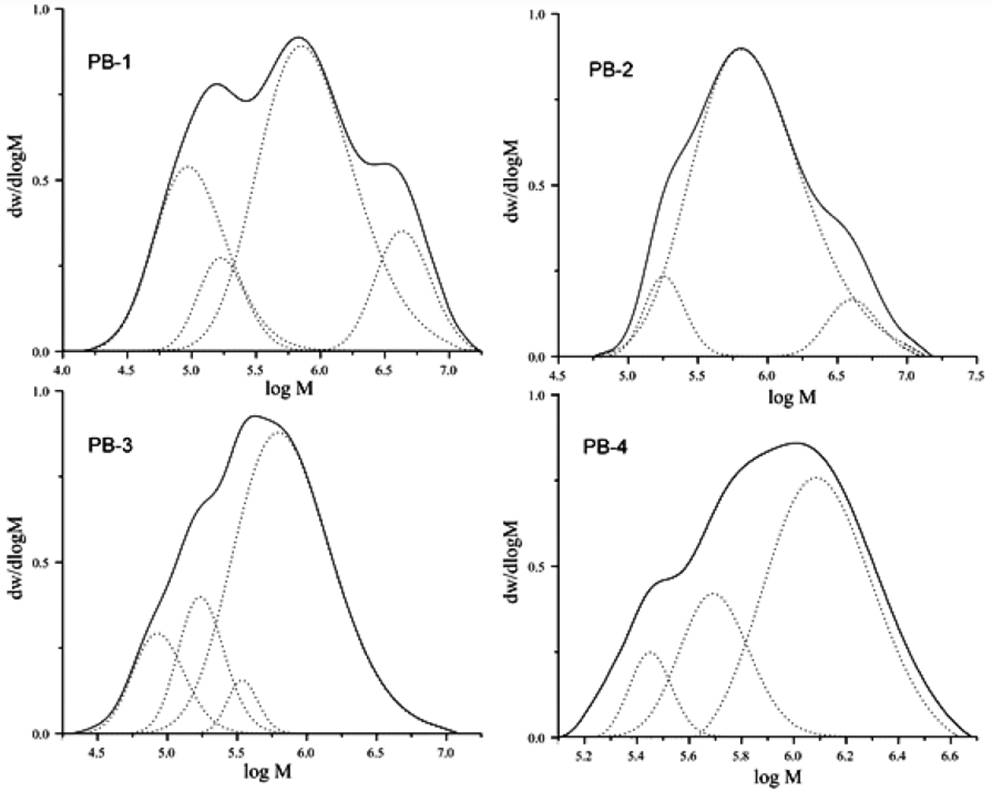
The deconvolution of the molar mass distributions, showed in Fig. 4, were carried out in order to give an approximate notion of the amount of types of active sites, elucidating the multimodal behavior of the employed catalysts, which exhibited the different types of active sites showed previously in Fig. 2, and which possess different probabilities of chain transfer and termination processes; therefore producing polymers with individual molar mass distribution and stereo-regularity, being the observed molar mass distributions (see Fig. 4) a superposition of all the normal distributions.
The microstructure of the polybutadienes on the other hand, standing by the disposition of double bonds along the polymer chain, was calculated by means of NMR and the values are shown in Table 1. In general the 1,4 structures content remained high (total cis + trans ≈ 98%) in all cases, so they are all in concordance to the mechanism of the bidentate coordination of the monomer, followed by the formation of an anti-allyl unit in the propagating polymer chain end, suggested by Friebe et al., [19]. TIBA and DIBAH prompted the highest cis-1,4 stereoregularity with 98.4 and 97.4% respectively; followed by TEA (91.8%) and lastly MAO which led to 90.5% cis-1,4 units. The 1,2-vinyl content remained very low in the case of all alkylaluminums as co-catalysts (< 1%), as expected, which indicates that the internal σ-allyl-metal bond is significantly unfavorable compared to the external σ-allyl-metal bond, which might be formed faster and more often than the internal σ-allyl bond [19, 20]. However the 1,2-vinyl content increased to 3.1% in the case of MAO as co-catalyst. This poor stereocontrol achieved with MAO, might be explained by the low propagation rate, reflected in the low % monomer conversion in function of time (see Fig. 3), and intensification of anti-sin isomerization role; since the initially formed anti allyl unit would lead exclusively to a cis-1,4 polymer if the rate of the propagation step with incoming monomers is faster than the rate of anti (cis) to syn (trans) isomerization. Furthermore, as it was mentioned before a possible coordination of the alcohol ligands to MAO, could have led to destabilize the active centers, decreasing the catalytic activity and definitely decrease the expected stereocontrol, even by occupying the vacancy of the Nd, available to monomer coordination, making the incoming monomer unit to coordinate with only one double bond and increasing the 1,2-vinyl content.
Regarding the thermal properties, it was not observed a significant difference by varying the co-catalyst (see Table 1), remaining the Tg values below to -107.1 °C, which are expected of high cis-1,4 polybutadienes [21].
Experimental
Reactants
All manipulations were carried out under inert argon atmosphere in an M-Braun glove box. Cyclohexane purchased from Aldrich was twice distilled from sodium under argon flow before use, and it was handled under inert atmosphere and stored in stainless steel containers. The anhydrous neodymium (III) chloride (NdCl3) (≥99.99%), TIBA 1.0 M in hexane, TEA 1.0 M in hexane, DIBAH 1.0 M in cyclohexane, MAO 10 wt.% in toluene, isopropanol (99.5%) and 1-pentanol (≥99%) were acquired from Aldrich and they were all used as received. 1,3-butadiene was purchased from Praxair and it was purified by passing it through activated molecular sieve 4Å and alumina.
Preparation and aging of neodymium-based catalyst
The catalyst system neodymium chloride tripentanolate of the general formula NdCl3·3L, where L = 1-pentanol was prepared by an alcohol interchange reaction between NdCl3 isopropanolate with 1-pentanol, as reported [10]. Later, in a glass container under inert atmosphere, the freshly prepared neodymium chloride tripentanolate was treated with a 1.0 M of the corresponding aluminum in solution at Al/Nd molar ratio of 35. The resultant mixture was stirred at 30°C for 15 minutes (aging step) prior the polymerization.
Polymerization procedure
Isothermal polymerizations were carried out in a 1 L stainless steel Parr reactor equipped with a turbine-type mechanical stirrer. The polymerization temperature was controlled by means of an electrical resistance and flow of cold water through an internal tubing coil operated by means of a PDI controller. The cyclohexane was introduced first and then1,3-butadiene monomer was added into the reactor through a 50 mL stainless steel container. After the monomer addition, the reactor was heated up to 50°C and the aged catalytic solution was fed into the reactor through a syringe, and setting-up the stirring at 100 rpm. The polymerizations were terminated by adding acidified methanol through a syringe, for later being precipitated in methanol, filtered and dried under vacuum at room temperature until constant weight. Irganox 1076 was added as stabilizer.
Characterization
The molecular weights of the resulting polymers were calculated by size exclusion chromatography (SEC) using a GPC Alliance 2690 from Waters, equipped with refractive index and viscometer detectors. Calibration was carried out with polystyrene standards and THF as eluent. The deconvolution of the molar mass distributions were carried out by means of the informatics software “peakfit”. The microstructure was determined by means of nuclear magnetic resonance (NMR) using a JEOL Eclipse-300 MHz spectrometer with deuterated chloroform as solvent and at room temperature. 1,2-vinyl isomer content in relation with 1,4- (cis + trans isomers) was calculated by 1H NMR, integrating the area of olefinic protons located in the chemical shift range from 4.8 to 5.7 ppm. The cis/trans ratio was calculated by 13C NMR (proton gated decoupling no-NOE experiments) integrating the area of aliphatic carbons located in the range from 27 to 33 ppm. Figures 5 and 6 show the 1H and 13C NMR spectra respectively corresponding to PB-2, in order to illustrate the assignment of signals. Differential scanning calorimeter (DSC) was performed by means of a TA instruments DSC 2920 at a heating rate of 10 °C/min and under inert atmosphere.


Conclusions
The polymerization of 1,3-butadiene by means of NdCl3 tripentanolate activated by TIBA, DIBAH, TEA and MAO was carried out. In general, all systems led to high cis-1,4 polybutadienes with multimodal molar mass distributions, characteristic of Nd based catalyst due to the multisite nature and chain transfer reactions to the co-catalyst.
Comparing the polymers formed with each activator, TEA was found to generate the highest polymerization activity but a rather low stereospecificty with 91.8% of cis-1,4 structure; and a relatively narrow molar mass distribution. TIBA on the other hand led to the highest cis-1,4 structure with 98.4%, as well as the narrowest molar mass distribution. DIBAH led to 97.4% of cis-1,4 structure and a relatively narrow molar mass distribution, however the catalytic activity and propagation rate was found to be lower than expected. MAO as co-catalyst on the other hand, let to the lowest polymerization activity and poorest stereo-control. By this we demonstrated the influence which each co-catalyst plays over the polymer final properties and which trend this influence follows, which is of sum importance when searching for a polymer with specific properties and applications.

