











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
Peptides are biomolecules whose long-standing interest among the scientific community has resulted in its wide and diverse application in various fields, spanning from drug discovery [1] to nanomaterials [2]. However the development of peptides as drug candidates is limited by their poor pharmacokinetic properties, their poor stability to enzymes in the gut and serum, and their poor absorption through the cell membranes; a partial remedy of these disadvantages can be achieved by the incorporation of N-methyl amino acids into peptides; which is a valuable strategy to improve their biological properties such as membrane permeability and proteolytic resistance, due to, alteration of the conformational characteristics or properties of the amide bond which induces conformational rigidity to the peptide backbone, which in turn blocks potential intramolecular hydrogen bonding sites and proteolytic enzyme cleavage sites [3,4]. Furthermore, N-Methyl amino acids represent important synthons for the design and synthesis of peptides with modified characteristics and therefore, may find application in drug discovery processes and structure activity relationship (SAR) studies [5]. N-methylation of amide nitrogen atoms increases the aqueous solubility of the peptide whilst simultaneously increasing the lipophilicity [6].
On the other hand, N-α-Methylation of amino acids causes steric hindrance during peptide coupling. Activated N-methylated amino acids are bulkier than their unsubstituted counterparts. The alkylated amino component may be more basic, but the slight gain in reactivity may be outweighed by the additional steric hindrance.
N-Methylation of the amide bonds in proteins is not a posttranslational modification mark. However, there are many N-methylated linear or cyclic peptides that have interesting biological functions. These peptides have demonstrated enormous potential as modulators of molecular functions. Also the N-methyl amino acids have been used in different systems to control the aggregation of peptides and proteins. For example, they have been used to regulate the aggregation of peptides in nanotubes [7], and block the dimerization of interleukin-8 [8].
The biochemistry of amyloid proteins has enriched the understanding of the assembling and folding dynamics of various proteins, as well as the pathogenic mechanism of several human diseases. For example, the increase of local concentration of Aβ peptides with the greatest fibrillogenic potential (particularly the Aβ-42) triggers the line of events causing Alzheimer´s disease (AD) [9,10,11], which, according to the amyloid cascade hypothesis [12], is initiated with the extracellular amyloid deposition of Aβ-peptides, essentially the Aβ-42. A potential goal in the prevention or therapy of AD is to decrease or eliminate neuritic plaques composed of fibrillar Aβ peptides, and it has been found that N-methyl amino acid-containing congeners of the hydrophobic “core domain” of Aβ inhibit the fibrillogenesis. A key feature of the inhibitor peptides is that they contain N-methyl amino acids in alternating positions of the sequence [13]. Based on these results, further research on the N-methylation of different residues to prevent the aggregation of Aβ peptides is needed/was carried out.
Thus, due to its high hydrophobic character, the 37-42 sequence (GGVVIA) of the Aβ-42 peptide could be prone to form fibrils itself as part of a peptide with a high tendency to fibrillation, although it is not known the exact contribution of this hexapeptide in the context of a 42-residue peptide. NMR studies [14] have suggested that the C terminus of the β strand binds to the segment Gly38 Ala42 to allow contact with the side chain of Phe19 and Met35 (Fig. 1). Also, computer simulations suggest that such sequence (GGVVIA) is crucial for the formation of amyloid fibrils. Furthermore, computational studies [15] propose that the sequence (GGVVIA) plays a major role in the generation of amyloid fibrils.
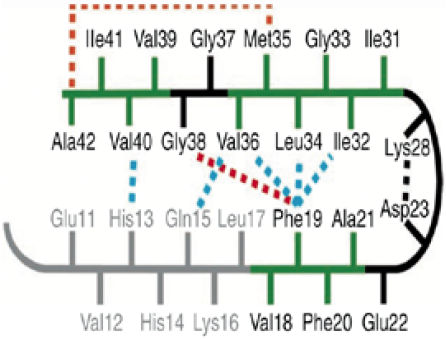
Phe19 and Gly38 (red dashed line) and between Met35 and Ala42 (orange dashed line). Taken from reference [14]
For the sequence GGVVIA it was calculated the fibrillation propensities using ZipperDB [15] database which contains predictions of fibril-forming segments within proteins identified by the 3D profile method, if the peptide structures have an energy threshold of -23 kcal/mol these segments with energies equal to or below this threshold are deemed to have a high fibrillation propensity. The sequence GGVVIA gives -27.800 kcal/mol therefore is predicted to form fibrils (Fig. 2).

In the present research the synthesis of the natural sequence 37-42 Aβ-42 (GGVVIA) is described, as well as the synthesis of two peptides with modifications in Val40 and Ile41, via the introduction of N-methyl amino acids. These modified peptides may find application as putative therapeutic agents for Alzheimer treatment and therefore the first challenge is the development of a synthetic route to prepare hydrophobic hexapeptides in solution-phase with efficient peptide couplings involving sterically hindered non-natural amino acids.
2. Results and Discussion
2.1 Retrosynthesis for sequence 37-42 (GGVVIA) of the peptide Aβ-42
The synthesis and purification of the sequence 37-42 (GGVVIA) of peptide Aβ-42 comprising mainly hydrophobic residues is challenging because of the low solubility in both hydrophilic and hydrophobic solvents and their high tendency to aggregate. Therefore, a convergent peptide synthesis in solution with a [3+3] fragment coupling strategy was chosen, which used the N-Boc/C-OBn strategy according to the retrosynthetic analysis shown in Scheme 1. This same strategy was employed to prepare the modified sequences: GGVV-NMeIA and GGV-NMeVIA. For the formation of the amide bond, the coupling reagents DCC (N, N´-dicyclohexylcarbodiimide) and HATU (N-[(dimethylamino)-1H-1,2,3-triazolo[4,5-b]pyridin-1 -yl-methylene)-N-methylmethanaminium hexafluorophosphate N-oxide) were tested, where better results were obtained by the latter. HATU has been proven to be very efficient in difficult sterically hindered couplings and usually gives a minimal level of racemization [16].

Scheme 1 Retrosynthesis for sequence 16 (Boc-GGVVIA-Bn), 17 (Boc-GGVV-NMeIA-Bn), and 18 (Boc-GGV-NMeVIA-Bn).
Numerous articles have described the synthesis, purification and analysis of α-peptides [17,18] where solid-phase synthesis seems to be preferred over solution phase because it affords automation of the assembly and circumvents multiple purification steps between each coupling. However, if large amounts of peptide are required, as for those used in therapeutic assays, solution-phase synthesis is usually needed.
Using the synthetic approach displayed in Scheme 2, the synthesis, optimization, characterization, and purification of the peptides, which were easily scaled, was carried out with good yields and low epimerization.

Scheme 2 Synthesis of peptides, (A) PhCH2OH, pTsOH, CHCl3, MW; (B) Boc2O, Et3N, MeOH, 50 ºC; (C) CH3I, NaH, THF, rt 24 h; (D) CH2Cl2, DIPEA, HATU, 0 ºC, 3-24 h; (E) HOBt, DCC, DIPEA, CH2Cl2, 24-72 h, rt; (F) Pd/C, H2, MeOH, rt 24 h; (G) CH2Cl2, TFA, rt 1-6 h.
Carboxylic acid protection is generally carried out through the formation of esters by reacting an acid with an alcohol in the presence of a catalyst at reflux [19]. However, the esterification of amino acids is more difficult than ordinary carboxylic acids, mainly because of zwitterionic structure [20,21]. Instead of the typical Dean-Stark procedure, the reaction was carried out using a microwave reactor, because reaction times were lower and the products were obtained without epimerization [22,23,24] according to specific rotation values reported, Scheme 2 (A).
The synthesis of the N-Boc residues was carried out using the usual protocol for the introduction of the the tert-butoxycarbonyl group (Boc) as shown in Scheme 2 (B), since it confers greater solubility in most common solvents and prevents or minimizes epimerization during coupling [25,26]. Commercially available N-methyl amino acids are still very expensive; therefore, various protocols have been developed for the synthesis of optically active N-methyl amino acids [27,28] and their incorporation into peptides [3,29]. Scheme 2 (C) shows the synthesis methodology of the N-methyl amino acids according to that reported by Malkov [30]. The yields of these reactions are indicated in Table 1, after purification by column chromatography.

As displayed in Scheme 2 (D, E), the synthesis of some peptides on Table 2 was carried out using HATU because with DCC the yields of the couplings with N-methyl amino acids were low and reaction times were more than 48 h. According to NMR spectra of 1H and 13C, no epimerization was observed.
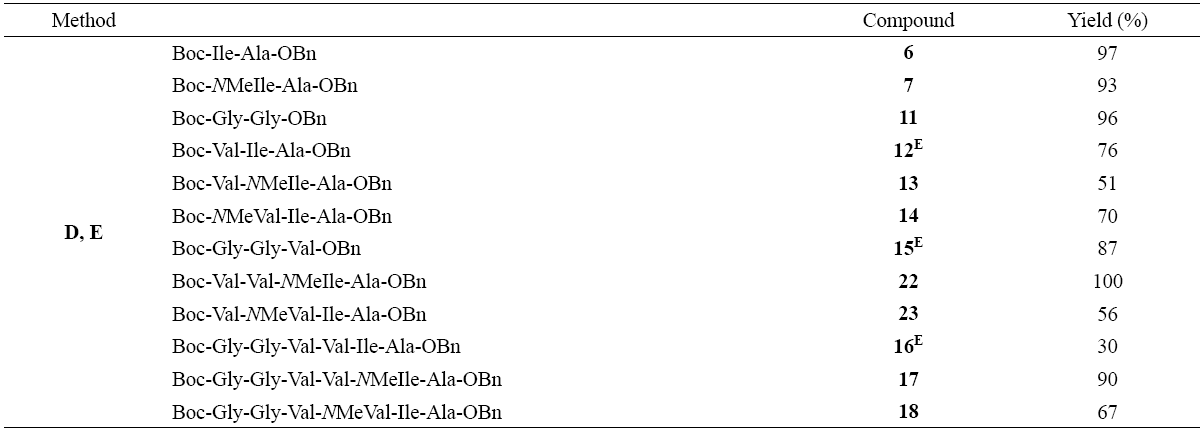
All peptides shown in Table 2 were purified by column chromatography and yields are indicated in Table 2. The removal of the Boc-protecting group was achieved using trifluoroacetic acid (TFA, method G) and immediately afterwards was coupled with the following residue. It should be noted that if this coupling was not done immediately, peptide yields significantly decreased down to 30%, due to the presence of the undesired 2,5-diketopiperazine (19) [31]. We found that a condition of pH = 8-9 was sufficient to achieve the cyclic product at room temperature, but if the salt of TFA is retained the product is stable and cyclization did not take place. Interestingly, when the peptide contains an N-methyl group, the cyclization reaction was not observed even at pH = 8-9 as displayed in the Scheme 3.

Scheme 3 Reaction conditions to favor the cyclic product 19[31] or linear peptide 20 and 21. (A) CH2Cl2, TFA, rt 1-6 h. (B) H2O, NaCO3, pH 8-9.
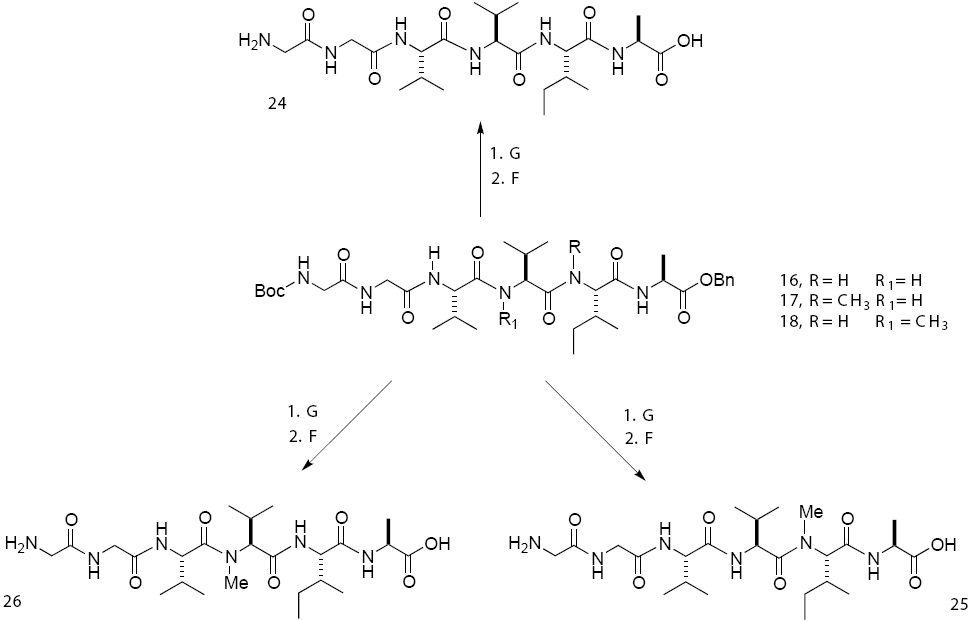
Due to poor performance of the above methodology in peptide synthesis Boc-Gly-Gly-Val-NMeVal-Ile-Ala-OBn (18), we developed a new synthetic sequence where tetrapeptides Boc-Val-Val-NMeIle-Ala-OBn (22) and Boc-Val-NMeVal-Ile-Ala-OBn (23) were first synthesized, followed by the removal of the Boc-protecting group and then coupled with dipeptide Boc-Gly-GlyOH [32].
The assignation of the NMR signals for each N-methylated compound used the data provided by COSY, HSQC or HMBC experiments, showing that the signals of alpha carbons presented different chemical shifts, depending on the residue position containing the N-methyl group.
Finally, the deprotection of the three hexapeptides was carried out first with TFA to remove the Boc-group, followed by the hydrogenolysis of the benzyl group (Scheme 4).
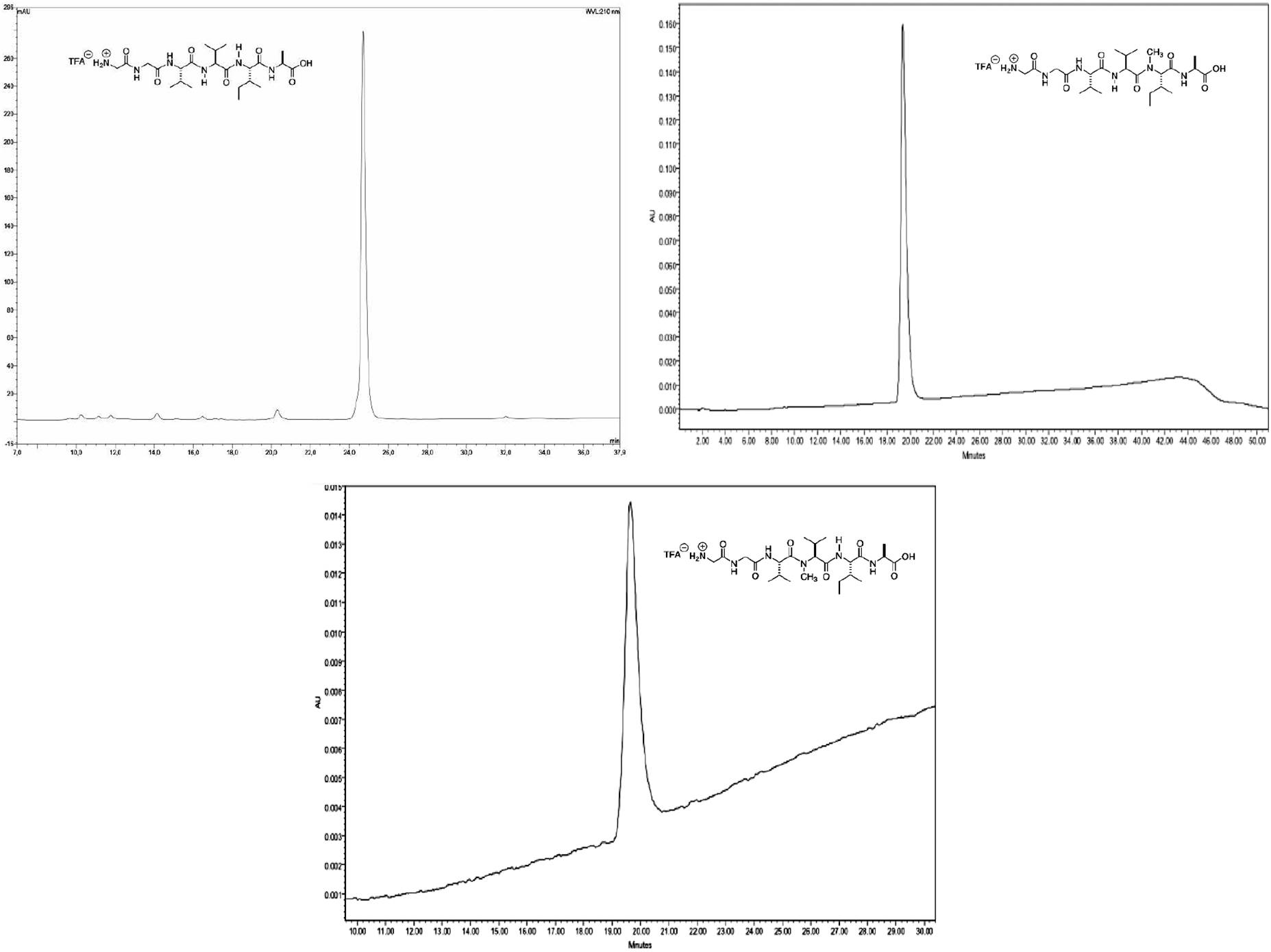
Purification of each peptide was performed by reverse phase flash chromatography. The chromatograms in Fig. 3 correspond to pure samples of RP-HPLC.
3. Experimental Section
3.1. General procedures
All chemicals and solvents of analytical grade were purchased by Sigma-Aldrich and used as received. All organic solutions were dried over Na2SO4. Thin layer chromatography was performed on aluminum plates coated with silica 60 F254 (Merck). Plates were visualized using UV light (254 nm) and 10% with ninhydrine in ethanol. Melting points were determined on a Buchi B-540 melting point apparatus and were not corrected. Optical rotations were obtained on a Perkin-Elmer 341 polarimeter at room temperature. Specific rotations ([α]D) are reported in deg/dm, and the concentration (c) is given in g/100 mL in the solvent specified. 1H and 13C NMR spectra were recorded on a Varian Gemini spectrometer at 200 and 50 MHz, Varian Oxford, at 400 and 100 MHz, respectively. The chemical shifts are given in ppm relative to tetramethylsilane, unless otherwise indicated. Various 2D-techniques and DEPT experiments were used to establish the structures and to assign the signals. Mass spectra were recorded on JEOL JMS-AX50SHA spectrometer. The reverse phase HPLC analyses were done using an Spherisorb ODS2 C18 (5 µm, 4.6*250 mm) column (for purity determination) with acetonitrile-water as mobile phase (gradient: 20-80% acetonitrile in 40 min).
3.2. General procedure for the synthesis of benzyl esters (A)
A mixture of the amino acid (1 equiv) with benzyl alcohol (3 eq), p-toluenesulfonic acid (1 equiv) and 15 mL of CHCL3 was added to an oven-dried 100-mL round-bottom flask equipped with a magnetic stir bar. The reaction was carried out in microwave reactor using an open system, a Dean Stark trap, 80-120 °C and an irradiation with 90-110 W for 1.5 h. The progress of the reaction was followed by TLC and visualized using UV light or by color development on spraying with ethanolic ninhydrine solution. The reddish reaction mixture was evaporated under reduced pressure to give a yellow-brown oil which was dissolved in water and brought to basic pH (8-9) with potassium bicarbonate and extracted (3x10 mL) with ethyl ether. The organic phase was dried over Na2SO4 and evaporated under reduced pressure leaving yellow oil, which was purified by column chromatography.
3.3. General procedure for the synthesis of urethanes (B)
An oven-dried 100-mL round-bottom flask equipped with a magnetic stir bar was charged with 1 equiv of the amino acid to be protected, MeOH and 1.2 equiv of Et3N. The reaction mixture was heated to 50 °C for 30 min and then allowed to be at room temperature, added 1.1 equiv of Boc2O, the solution was heated again at 50 °C, stirred for 2 h. After removal of solvent under reduced pressure and treatment with a solution of deionized water followed by acidification with citric acid to a pH of 2-4. The aqueous layer was extracted 3X10 mL of EtOAc. The layers were combined, dried over Na2SO4, and evaporated under reduced pressure, yielding a colorless oil.
3.4. General procedure for the synthesis of N-methyl-amino acids (C)
An oven-dried 100-mL round-bottom flask equipped with a magnetic stir bar was charged with solution of N-Boc-amino acid (1 equiv) and iodomethane (10 equiv) in anhydrous THF (5 mL/100 mg) under a stream of N2 and cooled (0 °C) then was added neat sodium hydride (10 equiv) slowly in portions over a period of 2 h. The reaction mixture was stirred at room temperature for 24 h under a nitrogen atmosphere and then diluted with ether (10 mL) and quenched with water (15 mL). The layers were separated, and the aqueous layer was extracted with ether (2 X15 mL), acidified to pH 3 with citric acid, and extracted with AcOEt (3 X20 mL). The combined organic phase was dried over Na2SO4 and evaporated to afford the corresponding N-methylated product as a thick colorless oil.
3.5. Coupling method (D)
The Boc-amino acid (or Boc-peptide) (1 equiv) was added to an oven-dried 100-mL round-bottom flask equipped with a magnetic stir bar, then was dissolved in CH2Cl2 (10 mL/100 mg), and DIPEA (4 equiv) was added. After stirring for 5 min at 0 °C, HATU (1 equiv) was added, followed by a solution of the benzyl ester of the amino acids (or peptides) (1 equiv). The reaction mixture was stirred in an ice bath for 3-6 h, and the mixture was evaporated and purified by column chromatography to give the desired product.
3.6. Coupling method (E)
HOBt (1.2 equiv) and a solution of the benzyl esters of amino acids (or peptides) (1 equiv) in CH2Cl2 (10 mL/100 mg) were added to a solution of the N-Boc derivatives of amino acids (or peptides) (1 equiv) and DIPEA (4 equiv) in CH2Cl2 at 0 °C. After stirring for 10 min, DCC (1.2 equiv) was added. The reaction mixture was stirred in an ice bath for 3 h, then at room temperature for 72 h, and the mixture was evaporated under reduced pressure until half volume, the solid was removed by filtration, and the filtrate was evaporated under reduced pressure. The mixture was purified by column chromatography to give the desired product.
3.7. General procedure for the removal of the Bn-protecting group (F)
A solution of the protected amino acid (or peptide) in methanol (5 mL/100 mg) was charged in an oven-dried 100-mL round-bottom flask equipped with a magnetic stir bar and hydrogenated in presence of palladium on carbon (10 wt %) a room temperature overnight. The reaction mixture was filtered and concentrated under reduced pressure obtaining the product.
3.8. General procedure for the removal of the Boc-protecting group (G)
The protected amino acid (or peptide) was added into an oven-dried 100-mL round-bottom flask equipped with a magnetic stir bar, and a 1:1 v/v trifluoroacetic acid (TFA):CH2Cl2 (1 mL/100 mg) mixture was added. The reaction mixture was stirred at room temperature for 6 h, and then concentrated under reduced pressure. Water was added to resulting solid and the solution was treated with potassium bicarbonate until basic pH (8-9). Then the mixture was extracted with AcOEt (3 x 10 mL), and the organic phase was concentrated and purified by column chromatography.
BocNMeIle-Ala-OBn (7): Obtained from compound (3) (245 mg) and (1) (179 mg) using the general coupling procedure (D), followed by usual work-up and purification. Compound (7) (378 mg, 93% yield) was isolated as a white solid; Mp 110-111 °C (from AcOEt), [α]D 25 = -101.5 (c 1.04, CHCl3), 1H-NMR (CDCl3, 400 MHz) δ = 0.77-0.85 [m, 6H], 0.94-0.99 [m, 2H], 1.30 [d, 3H, J = 6.8 Hz], 1.38 [s, 9H], 1.99 [br s, 1H], 2.71 [s, 3H], 4.06 [d, 1H, J = 11.2 Hz], 4.49-4.55 [m, 1H], 5.05-5.13 [m, 2H], 7.25-7.31 [m, 5H]. 13C-NMR (CDCl3, 100 MHz) δ = 10.6, 15.8, 18.2, 24.7, 28.5, 29.8, 30.4, 32.0, 48.1, 63.1, 67.2, 80.4, 85.2,128.3, 128.5, 128.7, 135.6, 157.0, 170.4, 172.5. HRMS (FAB) m/z = calcd for C22H34N2O5 406.2468; found: FAB+ [M+H]+ C22H35N2O5: 407.2551.
BocVal-Ile-Ala-OBn (12): Obtained from compound (6) (392 mg) by removal of the Boc-group using procedure (G), followed by coupling to compound (8) (217 mg) using the general coupling procedure (E), and usual work-up and purification. Compound (12) (373 mg, 76% yield) was isolated as a white solid: Mp 164-167 oC (from AcOEt), [α]D 25 = -62.5 (c 1.06, MeOH), 1H-NMR (CDCl3, 200 MHz) δ = 0.81- 0.92 [m, 12H], 1.09-1.15 [m, 2H], 1.37 [d, 3H, J = 7.4 Hz], 1.43 [s, 9H], 1.75-1.87 [br s, 1H], 1.94-2.01 [br s, 1H], 3.83-3.91 [br s, 1H], 4.22-4.30 [br s, 1H], 4.38- 4.49 [q, 1H, J = 7.2 Hz], 4.88 [s, 1H], 5.14 [s, 2H] 6.71 [d, 1H, J = 10 Hz], 6.81 [d, 1H, J = 6 Hz], 7.34 (br s, 5H], 13C-NMR (CDCl3, 50 MHz) δ = 11.4, 15.9, 17.5, 18.5, 19.9, 26.2, 28.8, 31.9, 34.9, 38.4, 58.7, 68.0, 80.6, 129.5, 129.5, 137.2, 158.0, 173.1, 173.6, 174.3. HRMS (FAB) m/z = calcd for C26H41N3O6: 491.2995, found: FAB+ [M+H]+ C26H42N3O6: 492.3034.
BocVal-NMeIle-Ala-OBn (13): Obtained from compound (7) (406 mg) by removal of the Boc-group using procedure (G), followed by coupling to compound (8) (217 mg) using the general coupling procedure (D), and usual work-up and purification. Compound (13) (257 mg, 51% yield) was isolated as a white solid: Mp 145-146 °C (from AcOEt), [α]D 25 = -103 (c 1.6, CHCl3), 1H-NMR (CDCl3, 400 MHz) δ = 0.81- 0.98 [m, 14H], 1.33 [d, 3H, J = 7.2 Hz], 1.42 [s, 9H], 1.89-1.98 [br s, 1H], 2.06-2.14 [br s, 1H], 3.07 [s, 3H], 4.37-4.41 [br s, 1H)], 4.54-4.60 [m, 1H], 4.72 [d, 1H, J = 11.6 Hz], 5.11-5.20 [m, 3H], 6.66 [d, 1H, J = 7.6 Hz], 7.31- 7.36 [m, 5H], 13C-NMR (CDCl3, 100 MHz) δ = 10.5, 15.6, 17.9, 18.1, 19.4, 24.4, 28.4, 30.9, 31.2, 31.6, 48.0, 55.6, 61.0, 67.3, 79.7, 128.3, 128.6, 128.7, 135.5, 156.1, 169.6, 172.4, 174.0. HRMS (FAB) m/z = calcd for C27H43N3O6: 505.3152, found: FAB+ [M+H]+ C27H44N3O6: 506.3180.
BocNMeVal-Ile-Ala-OBn (14): Obtained from compound (6) (392 mg) by removal of the Boc-group using procedure (G), followed by coupling to compound (9) (231 mg) using the general coupling procedure (D), and usual work-up and purification. Compound (14) (353 mg, 70% yield) was isolated as a white solid: Mp 100-101 °C (from AcOEt), [α]D 25 = -100 (c = 1.2, CHCl3), 1H-NMR (CDCl3, 400 MHz) δ = 0.83-0.93 [m, 12H], 1.09-1.13 [m, 2H], 1.39 [d, 3H, J = 6.8 Hz], 1.46 [s, 9H], 1.99 [br s, 1H], 2.23- 2.29 [br s, 1H], 2.78 [s, 3H], 4.05-4.08 [br s, 1H], 4.25-4.29 [br s, 1H], 4.59-4.63 [br s, 1H], 5.12-5.20 [m, 2H], 6.57-6.61 [m, 2H], 7.32-7.38 [m, 5H]; 13C-NMR (CDCl3, 100 MHz) δ = 11.3, 15.7, 18.5, 18.7, 19.2, 24.5, 26.0, 28.6, 30.2, 36.4, 48.3, 57.6, 64.9, 67.4, 80.6, 128.4, 128.6, 128.8, 135.5, 157.2, 170.6, 171.1, 172.6. HRMS (FAB) m/z = calcd for C27H43N3O6: 505.3152, found: FAB+ [M+H]+ C27H44N3O6: 506.3258.
BocGlyGlyValValNMeIleAlaOBn (17): Obtained from compound (13) (120 mg) by removal of the Boc-group using procedure (E), and from hydrogenolysis of BocGlyGlyValOBn (15) (100 mg) using the general removal procedure (F), using the general coupling procedure (D), and usual work-up and purification. Compound (17) (153 mg, 90% yield) was isolated as a white solid: Mp 68-69 °C (from AcOEt), [α]D 25 = -81.5 (c 1.2, MeOH), 1H-NMR (CDCl3, 200 MHz) δ = 0.74-0.91 [sa, 20H)], 1.24-1.34[br s, 3H)], 1.40 [s, 9H], 1.84-2.27 [br s, 3H)], 2.80 [s, 3H], 3.20 [br s, 2H)], 3.83 - 4.00 [br s, 2H)], 4.09-4.25 [br s, 1H)], 4.51-4.67 [br s, 1H)], 4.73-4.88 [br s, 2H)], 5.04-5.19 [sa, 2H)], 5.97 [s, 1H], 7.26-7.34 [br s], 7.68 [br s, 2H], 7.86-7.89 [br s, 1H], 8.39-8.43 [br s, 1H]. 13C-NMR (CDCl3, 175 MHz) δ = 10.7, 12.4, 15.1, 17.3, 18.5, 18.5, 18.6, 18.8, 24.6, 28.3, 31.1, 31.7, 32.0, 32.2, 38.5, 42.7, 43.7, 47.6, 53.9, 55.7, 57.7, 60.5, 67.0, 79.9, 128.0, 128.1, 128.3, 128.5, 135.3, 156.2, 168.5, 169.9, 170.1, 171.0, 173.2.
BocGlyGlyValNMeValIleAlaOBn (18): Obtained from compound (20) (120 mg) by removal of the Boc-group using procedure (G), and from hydrogenolysis of BocGlyGlyOBn (11) (63 mg) using the general removal procedure (F), using the general coupling procedure (D), and usual work-up and purification. Compound (18) (95 mg, 67% yield) was isolated as a white solid: Mp 69-70 °C (from AcOEt), [α]D 25 = -88.8 (c 1, MeOH), 1H-NMR (CDCl3, 200 MHz) δ= 0.73- 0.92 [br s, 20H)], 1.38 [d, 3H, J = 6.8 Hz], 1.43 [s, 9H], 1.88-2.02 [br s, 2H], 2.17- 2.23 [br s, 1H], 3.21 [s, 3H], 4.19-4.24 [m, 1H], 4.33-4.38 [m, 1H], 3.79- 3.81 [br s, 2 H], 3.92 [d, J= 4.2 Hz, 1H], 4.08 [d, 1 H, J = 4.6 Hz], 4.16-4.29 [br s, 1H], 4.58-4.66 [br s, 1H], 4.73-4.81 [br s, 1H], 4.87-4.93 [br s, 1H], 5.12 [d, 2H], 5.48 [br s, 1H], 7.07-7.11 [br s, 2H], 7.30-7.31 (br s, 5H], 8.16-8.29 [br s, 2H]. 13C-NMR (CDCl3, 50 MHz) δ = 10.3, 12.2, 17.9, 18.1, 18.6, 18.8, 19.0, 21.2, 24.6, 26.8, 28.2, 31.0, 31.3, 31.4, 35.6, 39.8, 42.3, 43.9, 48.1, 54.4, 57.4, 62.1, 67.0, 80.05, 128.4, 128.6, 128.8, 135.4, 156.1, 168.7, 169.7, 170.8, 171.2, 172.5, 173.3.
BocVal-Val-NMeIle-Ala-OBn (22): Obtained from compound (13) (253 mg) by removal of the Boc-group using procedure (G), followed by coupling to compound (8) (109 mg) using the general coupling procedure (D), and usual work-up and purification. Compound (22) (299 mg, 99% yield) was isolated as a white solid: Mp 93-94 °C (from AcOEt), [α]D 25 = -80.3 (c 1.32, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ = 0.79- 0.94 [m, 20H)], 1.30 [d, 3H, J = 7.2 Hz], 1.42 [s, 9H], 1.89-1.94 [br s, 2H], 2.06- 2.13 [br s, 1H], 3.17 [s, 3H], 4.19-4.23 [br s, 1H], 4.54-4.62 [m, 1H], 4.73-4.77 [m, 1H], 4.8 [d, 1H, J = 11.2 Hz], 5.11-5.21 [m, 3H], 7.28- 7.34 [m, 5H], 7.46 [d, 1H, J = 7.2 Hz], 7.7 [d, 1H, J = 6.8 Hz]; 13C-NMR 10.7, 15.2, 17.4, 18.0, 18.4, 18.8, 24.5, 28.3, 30.9, 31.3, 32.0, 47.6, 54.1, 58.9, 60.5, 67.0, 79.5, 128.1, 128.5, 135.3, 155.6, 169.8, 171.7, 172.7, 173.2, HRMS (FAB) m/z= calcd for C32H52N4O7: 604.3836, found: FAB+ [M+H]+ C32H53N4O7: 605.3890.
BocVal-NMeVal-Ile-Ala-OBn (23): Obtained from compound (14) (253 mg) by removal of the Boc-group using procedure (G), followed by coupling to compound (8) (109 mg) using the general coupling procedure (D), and usual work-up and purification. Compound (23) (169 mg, 56% yield) was isolated as a white solid: Mp 145-146 °C (from AcOEt), [α]D 25 = -103.5 (c 1.6, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ = 0.76- 0.94 [m, 20H], 1.35 [d, 3H, J = 7.2 Hz], 1.38 [s, 9H], 1.86-1.92 [br s, 1H], 1.97-2.02 [br s, 1H], 2.21-2.30 [br s, 1H], 3.16 [s, 3H], 4.19-4.24 [m, 1H], 4.33-4.38 [m, 1H], 4.58-4.65 [m, 1H], 4778 [d, 1H, J= 11.2 Hz], 5.10-5.18 [m, 2H], 5.77 [d, 1H, J = 9.6 Hz], 6.60 [d, 1H, J = 7.2 Hz)], 7.29- 7.35 [m, 5H], 7.43 [d, 1H, J = 8.8 Hz], 13C-NMR (CDCl3, 100 MHz) δ = 10.7, 15.3, 18.0, 18.2, 18.3, 24.6, 26.4, 28.2, 30.8, 30.9, 35.8, 47.9, 55.7, 57.4, 62.3, 67.1, 79.2, 128.1, 128.3, 128.5, 135.2, 156.1, 170.5 (2 C = O), 172.4, 174.0, HRMS (FAB) m/z = calcd for C32H52N4O7: 604.3836, found: FAB+ [M+H]+ C32H53N4O7: 605.3890.
TFA-GlyGlyValValNMeIleAlaOH (25): Obtained from compound (17) (153 mg) by removal of the Boc-group using procedure (G), and from hydrogenolysis using the general removal procedure (F), and usual work-up and purification. Compound (25) (112 mg, 90% yield) was isolated as a white solid: Mp 198-199 °C (from MeOH), [α]D 25 = -145 (c 1.1, H2O), 1H-NMR (CD3OD, 400 MHz) δ = 0.83-0.99 [br s, 21H)], 1.27-1.36[br s, 4H)], 2.00-2.13 [br s, 3H)], 3.16 [s, 3H], 3.76 [s, 2H)], 4.01 [s, 2H)], 4.16-4.25 [br s, 1H)], 4.34-4.41 [br s, 1H)], 4.47 [d, 1H, J = 6.8 Hz], 4.57 [d, 1H, J = 9.2 Hz], 4.66-4.69 [br s, 2H)], 7.81 [d, 1H, J = 6.8 Hz], 8.17 [d, 1H, J = 8.8 Hz], 8.45 [d, 1H, J = 8.4 Hz]. 13C-NMR ((CD3OD, 100 MHz) δ = 9.4, 9.8, 14.3, 17.4, 17.6, 17.8, 18.3, 24.3, 30.3, 30.6, 31.0, 31.7, 40.4, 41.9, 49.7, 54.5, 58.6, 60.8, 173.4, 173.0, 172.1, 169.4, 167.0, 167.1. HRMS (ESI) m/z = calcd for C24H44N6O7: 528.32, found: ESI+ [M+H]+ C24H45N6O7: 529.3342.
TFA-GlyGlyValNMeValNMeIleAlaOH (26): Obtained from compound (17) (95 mg) by removal of the Boc-group using procedure (G), and from hydrogenolysis using the general removal procedure (F), and usual work-up and purification. Compound (26) (62 mg, 90% yield) was isolated as a white solid: Mp 189-190 °C (from MeOH), [α]D 25 = -145.5 (c 1, H2O); 1H-NMR (D2O, 400 MHz) δ = 0.66-0.85 [br s, 18H)], 0.99-1.07 [br s, 1H)], 1.23[d, J = 7.2 Hz 3H)], 1.26-1.36 [br s, 1H)], 1.70-1.76 [br s, 1H)], 1.88-1.97 [br s, 1H)], 2.05-2.14 [br s, 1H)], 3.07 [s, 3H], 3.71 [s, 2H)], 3.88 [d, 1H, J = 5.6 Hz], 3.93 [d, 1H, J = 5.6 Hz], 3.97-4.10 [br s, 4H)], 7.96 [d, 1H, J = 6.8 Hz], 8.13 [d, 1H, J = 7.6 Hz], 8.25 [d, 1H, J = 8.4 Hz], 8.38-8.41 [br s, 1H)]. 13C-NMR (D2O, 100 MHz) δ = 9.6, 14.6, 17.4, 17.5, 17.7, 18.1, 18.2, 24.4, 26.2, 30.1, 30.9, 35.6, 40.6, 42.0, 50.7, 55.6, 58.2, 62.6, 167.7, 170.7, 171.8, 172.2, 174.3, 179.0. HRMS (ESI) m/z = calcd for C24H44N6O7: 528.32, found: ESI+ [M+H]+ C24H45N6O7: 529.3344.
4. Conclusions
The solution phase synthesis of peptides containing N-methylamino acids constitutes a real challenge. Herein, we describe an approach that allows for efficient coupling of N-methyl amino acids to other units, including other N-methyl residues, maintaining stereochemical integrity. The process, which uses HATU as coupling reagent, takes place in high yields and short reaction times, under mild reaction conditions (room temperature) compared with DCC, which need more reaction time (24-72 h) and gives lower yields. Afterwards, the difficult sequence 37-42 (GGVVIA) of Aβ42 was synthesized, characterized and modified by the addition of NMe groups on Val40 and Ile41 obtaining the sequences GGVNMeVIA and GGVVNMeIA. From ZipperDB, the sequence GGVVIA gives -27.800 kcal/mol therefore is predicted to form fibrils; thus N-methylated peptides coul be tested for inhibition of fibrillation of the natural sequence.

