











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Porous coordination polymers (PCPs, also known as metal-organic frameworks or MOFs) are a promising class of porous materials constructed from metal ions and organic ligands. Thanks to their structural stability and functional flexibility, the field of PCPs has become one of the fastest growing research areas in chemistry. After many years of research and a outwardly ever expanding literature dedicated to PCPs [1] it is actually hard to remember the days when journals were not crowded with articles devoted to these stylish, even fancy, three-dimensional framework structures [2]. During the 90's such structures were still uncommon, even though coordination polymers had been reported as far as 1916 [3] and the remarkable work of Robson [4-6] had proposed the methodology that would go on to nature the larger field. The current relevance of this area [7] is dependent upon the important contributions by Kitawaga [8], Yaghi [9] and others, in particular the identification of the properties that PCPs exhibit. Nevertheless, before all these major advances, researchers were drawn in by the fascination and beauty of coordination polymers.
Coordination Polymers
Polymers are defined as high molecular weight materials formed by repetition of monomeric units linked with covalent bonds [10]. Coordination polymers [11-16] are infinite systems built from metal ions and organic ligands as the building blocks, which are linked by coordination bonds and other weak chemical bonds. As mentioned before, these compounds are also known as metal-organic frameworks (MOFs) [13]; or metal-organic coordination networks (MOCNs) [13]. The solid state structures of crystalline coordination polymers can be determined by single X-ray diffraction methods which confirm that coordination polymers have structural diversity and porosity. The development of coordination polymers in materials science, crystal engineering and supramolecular chemistry are areas of research undergoing significant growth [14, 17-26]. The design and synthesis of such materials is driven by their potential in a diverse range of applications such as gas storage [27, 28], catalysis [29], electrical conductivity [30], luminescence [31], magnetism [32, 33] and optics [34].
The process of building a coordination polymer is mainly directed by the coordination bonds that are defined [10] either by the donation of a lone electron pair from the ligand (Lewis base) to the metal cation (Lewis acid) or by electrostatic attractions between positively charged metal ions and a negatively polarised or charged donor ligand. The energy of such interactions is usually between 50 and 200 kJ mol-1[35]. However, coordinate and covalent bondings are not the only interactions present in these compounds. Weaker non-covalent interactions [36] such as hydrogen bonds, van der Waals forces and π-π contacts may also strongly influence the formation of coordination polymers.
The arrangement of the components of coordination polymers mostly exists in the solid state, thus, these components interact, in solution, through coordination interactions and weaker forces (non-covalent interactions) thus, the coordination polymers grow through self-assembly [36 37] (Fig. 1). Self-assembly is the organisation of molecules into larger structures through non-covalent interactions to form for example micelles, membranes, vesicles and liquid crystals [36, 38]. Also, the role of the non-covalent interactions is important in the packing of the one-dimension chains, two dimension nets and three-dimensional frameworks [39, 40].
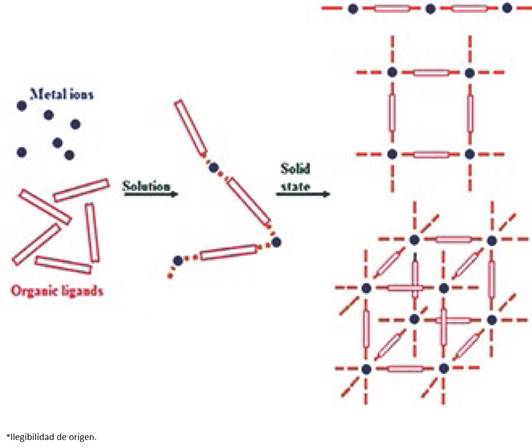
Fig. 1 Schematic illustrating the formation of coordination polymers from their building blocks. (Adapted from Ref [41])
Connectors and Linkers
Coordination polymers include two essential components, connectors (metal ions) and linkers (ligands) [15], and these can be defined as the building blocks components. In addition, there are other components such as counterions and solvent molecules (non-bonding guests) that must be considered [42]. Counterions can influence the metal ion environment and also the overall structure through weak interactions or through their presence in void spaces within the coordination polymer. Solvent molecules, present during the initial synthesis, often co-crystallise with the coordination polymer increasing the number of possible interactions in the solid state [42]. These solvent molecules may be present as guest molecules in the vacant spaces within the polymer structure and they may play a crucial role in the construction of highly porous materials [43].
The fundamental characteristics of both connectors and linkers are the coordination numbers and coordination geometries of the components. From these we can understand both the number and orientation of their respective binding sites [15].
Transition-metal ions are known to be versatile connectors in the construction of coordination polymers. Depending on the metal ion, and its oxidation state, coordination numbers usually vary from 2 to 7 providing various geometries such as linear, trigonal, tetrahedral, square planar, square pyramidal, trigonal bipyramidal, octahedral, trigonal prismatic, pentagonal bipyramidal and distorted forms of these geometries [42]. The polyhedral coordination geometries of the lanthanide ions with coordination numbers ranging from 7 to 10 may be useful for the generation of new and atypical network topologies [44].
Organic ligands are utilised as versatile linkers in the synthesis of coordination polymers and act as bridging groups between the metal ions [14]. For infinite expansion, organic ligands must be multidentate with at least two donor atoms for instance N-, Oor S-donors [12]. These linkers may differ from each other in their charges, and the majority of ligands used are neutral or anionic [14]. Others factors that may influence the structure of coordination polymers are the shape, length and the functionalities present on the ligand [15]. The most frequently used neutral organic ligands are pyrazine (pyz) and 4.4'-bipyridine (4,4'-bpy) [39, 45-47]. An example of a coordination polymer formed from 4,4'-bpy and a metal ion (M) is shown in Fig. 2 [48]. Polycarboxylate ligands such as di[49-54], tri[49, 51-54], tetra[55-58], and hexacarboxylates [59-62] are typical anionic linkers.
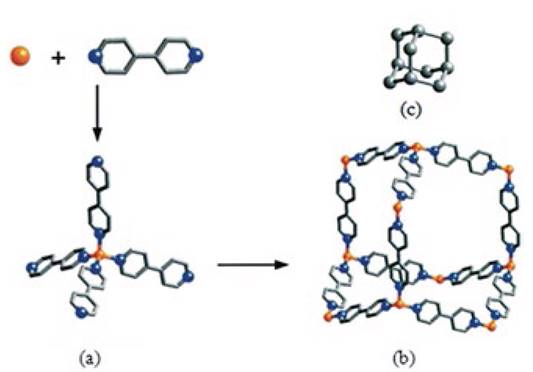
Fig. 2 Assembly of a coordination polymer by (a) the co-polymerisation of metal ions with organic: linkers to give, (b) a flexible metal(4,4'-bpy) structure that shows (c) expanded diamond topology. Metal ion (M) orange, C gray and N blue (Reprinted with permission from Eddaoudi, M.; Moler, D. B.; Li, H. L.; Chen, B. L.; Reineke, T. M.; O'Keeffe, M.; Yaghi, O. M. Acc. Chem. Res. 2001, 34, 319-330.
Dimensionality
Coordination polymers can extend infinitely in one(1D), two(2D) or threedimensions (3D). The ligand (organic group) must be a bridge, in at least one dimension [14, 15], and the metal ions must be linked by this organic ligand. Thus, at least one carbon atom must lie between one donor atom and the next.
Fig. 3 shows examples of the connectivity between metal ions and ligands. Dimensionality is often determined by the metal centres. In one-dimensional materials the metal ion is coordinated with two ligand molecules, metal ions and organic ligands alternate infinitely, leading to a chain [14]. Two-dimensional compounds are obtained when three or four ligands coordinate to the metal ion and expand in two directions leading to sheets [15]. With metal ions of higher coordination number three-dimensional structures can be built [14, 15, 42]. The elementary units used are not always so simple and 1D, 2D or 3D architectural types are possible depending on the connectors and linkers used and the experimental conditions.
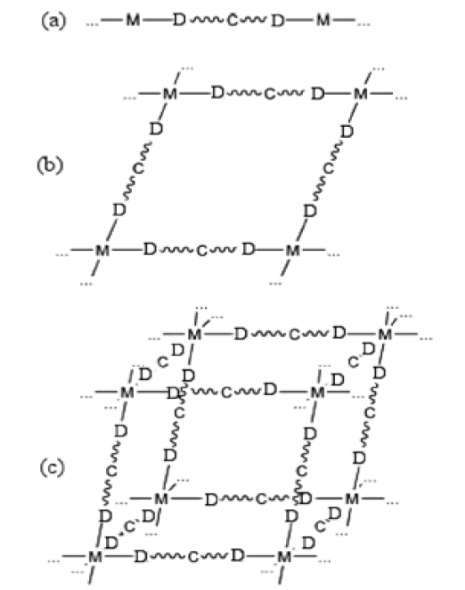
Fig. 3 Representation of coordination polymer linking ligands with one carbon atom (C) in-between the donor atoms (D) and the metal centre (M). Donor atoms (D) can be oxygen, sulphur, nitrogen, phosphorous, etc. (a) one-dimensional (1D), (b) two-dimensional (2D), (c) three-dimensional (3D) structures (Reproduced from Ref [14])
One-dimensional Coordination Polymers
One-dimensional coordination polymers normally exist as one of seven types shown in Fig. 4 [63-67], and the packing mode of these chains determines whether porosity is created. If the pores are blocked by other chains structures, little or no free space will be formed. Holman et al. [68] synthesised the coordination polymer {[Co(H2O)4(pyz)(NO3)2]·2H2O}∞ , (pyz = pyrazine) which has a one-dimensional linear chain structure of alternating pyrazine molecules and Co(H2O)4 units. Cobalt ions are hexa-coordinated with the O atoms from four coordinated H2O molecules occupying the equatorial positions and the N atoms of two different pyrazine ligands present in the axial positions.
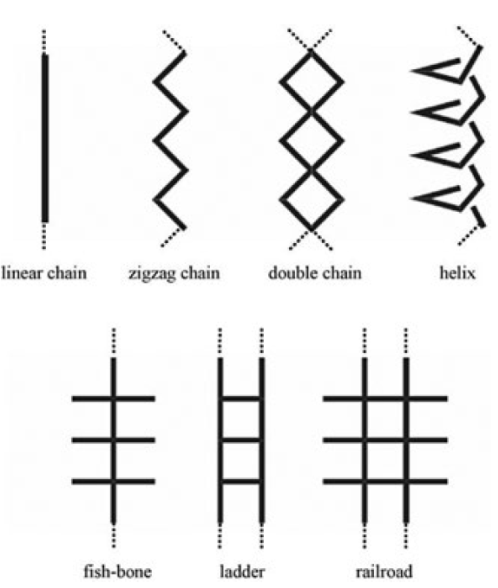
Fig. 4: One-dimensional coordination polymer assemblies (Kitagawa, S.; Kitaura, R.; Noro, S-. I. (2004), Functional Porous Coordination Polymers. Angew. Chem. Int. Ed., 43 (18).
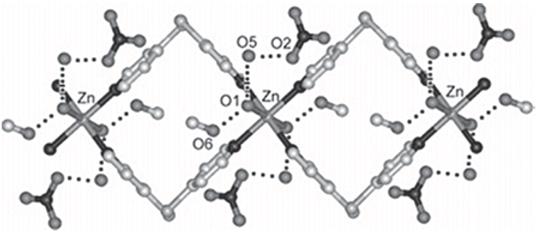
Horikoshi and co-workers reported [69] the Zn(II) complex {[Zn(H2O)2(4dps)2.(NO3)2 (H2O)(CH3OH)]}∞, (4dps = 4,4'-dipyridyl disulfide). This complex exhibits a one-dimensional double chain structure (Fig. 5) unit Zn(II) hexa-coordinated with the equatorial positions occupied by four N atoms from four different ligand molecules and by two H2O molecules in the axial positions.
Cheng et al. synthesised [70] the one-dimensional helical chain {[HgBr2(2,5-bis(3-pyridil)-1,3,4-oxadiazole)]}∞ in which the Hg(II) ion has a distorted tetrahedral environment consisting of two N atoms from two distinct ligands and two terminal bromide atoms. The HgBr2 unit and the ligands alternate to form a one-dimensional chain. The bent shape of the ligand molecules and its coordination made at the Hg(II) centre induces a helical twist to the one-dimensional chain.
Two-dimensional Coordination Polymers
Two-dimensional coordination polymers are usually found in one of the eight forms presented in Fig. 6 [71-75]. One of the most powerful techniques for the analysis and design of coordination polymers is to reduce their crystal structures to networks (or nets). A network is a polymeric collection of interlinked nodes where each link connects two nodes and each node is linked to three or more other nodes [13]. Networks can help the description and understanding of complicated structures more easily and provide evidence for a particular packing arrangement and any corresponding properties [44].
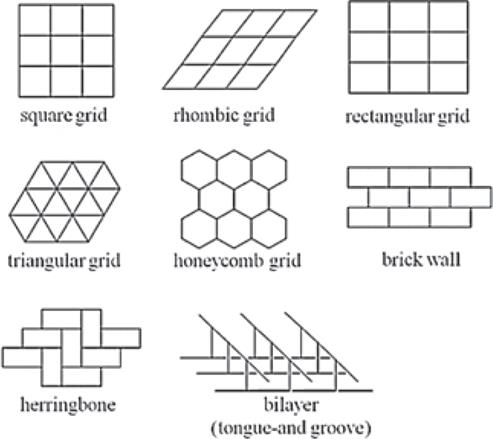
Fig. 6 Two-dimensional coordination polymer assemblies (Kitagawa, S.; Kitaura, R.; Noro, S-. I. (2004), Functional Porous Coordination Polymers. Angew. Chem. Int. Ed., 43 (18).
Square grid networks are the simplest example of two-dimensional coordination polymers [12]. In this example the proportion of metal to ligand usually is 1:2 with the metal centres coordinated to four different ligand molecules. Repetition of this unit allows propagation of the structure in two dimensions [46]. Uemura and co-workers reported [76] the network {[Co(SCN)2(3-pna)2]}∞, (SCN = thiocyanate), (3-pna = N-(3-pyridyl)nicotinamide). The crystal structure of this coordination polymer shows a two-dimensional sheet composed of a square grid (Fig. 7).
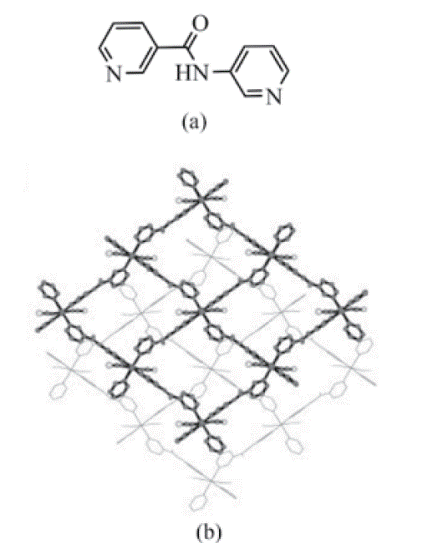
Fig. 7 (a) View of the the ligand N-(3-pyridyl) nicotinamide; (b) two-dimensional sheet structure of {[Co(SCN)2(3-pna)2]}∞ (SCN = thiocyanate), (3-pna = N-(3-pyridyl)nicotinamide) (Uemura, K.; et al (2002), Novel Flexible Frameworks of Porous Cobalt(II) Coordination Polymers That Show Selective Guest Adsorption Based on the Switching of Hydrogen-Bond Pairs of Amide Groups. Chem. Eur. J., 8 (16).
Where metal ions are only coordinated by three ligand molecules in a T-shape layers are constructed resulting in either honeycomb grid, brick wall, herringbone or bilayer structures (Fig. 6). Spichal et al. reported [77] two-dimensional coordination polymers containing lanthanide ions of the general formula {Ln(NO3)3(dppeO2)1.5}∞, (Ln = Pr, Nd, Sm, Dy) (Ph2P(O)-CH2CH2-P(O)Ph2 = dppeO2) (Fig. 8). In these examples the ratio of metal to ligand is 2:3 and in order to produce T-shaped connectors some coordination sites of the metal centre must be blocked by counter anions, such as halides or nitrates or by terminal ligands.
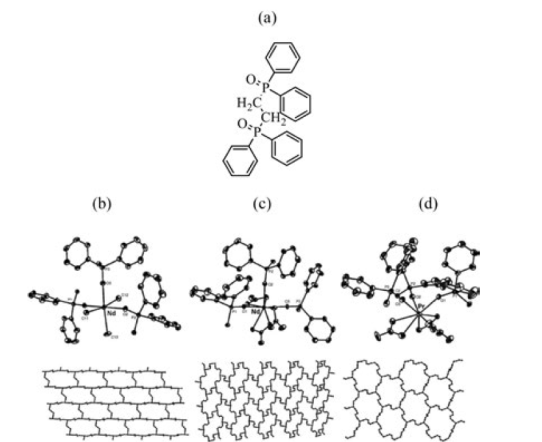
Fig. 8 (a) View of the ligand Ph2P(O)-CH2CH2-P(O)Ph2 = dppeO2; (b) brick wall assembly in {[NdCl3(dppeO2)1.5]}∞, (c) parquet floor assembly in {[Nd(NO3)3(dppeO2)1.5]}∞ and (d) pseudo-honeycomb assembly in {[Pr(NO3)3(dppeO2)1.5]}∞ (Adapted with permission from Spichal, Z.; Necas, M.; Pinkas, J. Inorg. Chem. 2005, 44, 2074-2080.
Three-dimensional Coordination Polymers
Three-dimensional intersecting channels, commonly found in zeolites [78, 79], are constructed by the interconnection of one-dimensional chains in various directions [78, 80]. Various types of three-dimensional networks have been reported for coordination polymers [49] and topological analysis of these networks shows common three-dimensional nets such as diamond, PtS, cubic or NbO structures (Fig. 9) [49]. The nodes in these frameworks have either tetrahedral or octahedral connectivity and it is relatively easy to analyse these three-dimensional networks through the mononuclear metal centres and linear bidentate ligands [49, 81].
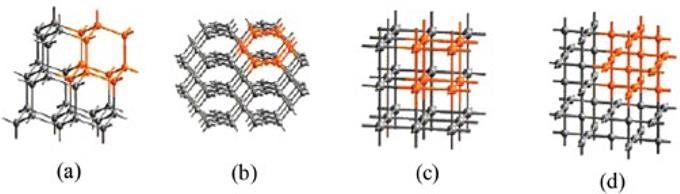
Fig. 9 Examples of common three-dimensional nets (a) diamond, (b) PtS (c) cubic and (d) NbO (Reprinted with permission from Eddaoudi, M.; Moler, D. B.; Li, H. L.; Chen, B. L.; Reineke, T. M.; O'Keeffe, M.; Yaghi, O. M. Acc. Chem. Res. 2001, 34, 319-330.
For example, in the diamond network [46, 83], each node is connected to four bridging ligands in a tetrahedral geometry, leading to a three-dimensional diamond-like network. Chen and co-workers reported [84] the framework {[Cd(3,3'AZDB)2].(H2NMe)2 (NH4)} ]}∞, (3,3'-AZDB = 3,3'-azodibenzoate) (Fig. 10). In this structure the Cd(II) ions are eight-coordinate but can be considered to be nodes with four connections to bidentate carboxylate groups.
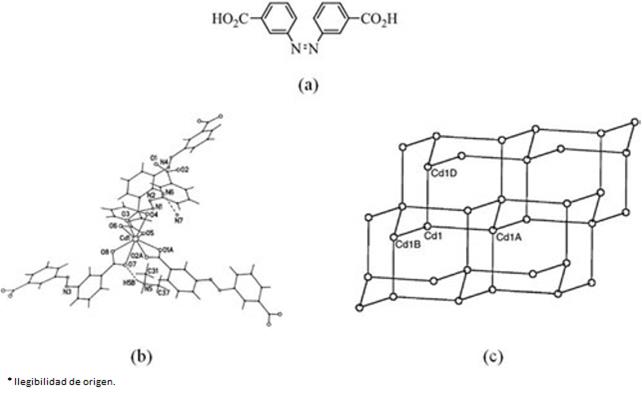
Fig. 10 (a) View of the ligand 3,3'-azodibenzoic acid = 3,3'-H2AZDB, (b) eight-coordinate Cd(II) in {[Cd(3,3'-AZDB)2].(H2NMe)2(NH4)}∞, 3,3'-AZDB = 3,3'-azodibenzoate and (c) schematic representation of the diamondoid network in {[Cd(3,3'-AZDB)2]·(H2NMe)2 (NH4)}∞ (Adapted from Ref [83])
Synthesis of Coordination Polymers
One of the challenges in the synthesis of coordination polymers is to obtain single crystals suitable for detailed crystallographic analysis. Unlike molecular species, most coordination polymers are insoluble once synthesised and therefore the preparation of crystals by recrystallisation is often not an option [15]. Usually, where a network can be dissolved it is only through the use of polar solvents, a strong acid or base, which will break up the polymer and then be incorporated in the recrystallised species resulting in a different material to the original phase [85]. Therefore, crystals of coordination polymers are normally directly obtained from synthetic reactions, although single crystals can be obtained from mixed solutions, often it is crucial to use slow precipitation methods to obtain high quality crystals [86]. It is important to note that the structures obtained from single crystals may be not representative of the bulk material which is tested to determine the coordination polymer properties [16]. Furthermore, reactions often result in more than one product. Thus, it is essential to ensure a correlation between the single crystals and the bulk product, and commonly this is achieved via the use of X-ray powder diffraction analysis of the bulk material.
There are four main synthetic strategies for the generation of crystalline coordination polymers reported in the literature [48 87]. The saturation method results in the formation of crystals from a mixture of the different reagents. The diffusion method is based upon slow contact of the different species. This method preferred as it often results in crystals suitable for X-ray diffraction analysis.
Hydro(solvo)thermal methods take advantage of the self-assembly of products from soluble precursors. The operating temperature range is usually 120-260 °C inside a sealed container (autoclave) under autogenous pressure [85]. The difference in solubility between organic and inorganic components in a common solvent is often a barrier to the formation of single crystals [48]. Thus, solvothermal experiments present an attractive alternative approach as the solubilities of starting materials may be increased [88]. Microwave methods [89] can also be used to improve the solubilities of the reagents in a solvent in order to facilitate the reaction and achieve crystalline products [13].
Outline of Microporous Properties
Coordination polymers have diverse potential applications in gas storage [27, 28], catalysis [29], electrical conductivity [30], luminescence [31], magnetism [32, 33], and optics [34]. However, in order to obtain the full potential of these materials, the access to the pores is fundamental. Therefore, a description of the microporosity of coordination polymers will now be presented.
Pores are defined as small openings through which fluids or gases can pass [90]. Thus, for a material to be termed porous, confirmation of the porosity beyond structural analysis must be demonstrated. Normally this is achieved by gas adsorption-desorption studies [90] which can provide direct evidence of permanent porosity. Permanent porosity is defined as the retention of structural integrity during the adsorption and desorption of guest species from the host material (Fig. 11) [15, 28]. It may also be demonstrated directly through powder or single crystal X-ray diffraction studies, but generally porosity is confirmed by gas sorption studies [90].
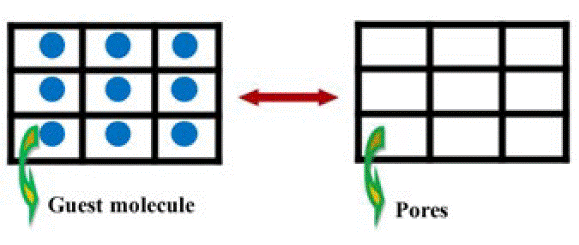
Fig. 11 Schematic representation of permanent porosity where the guest molecules located in the pores of the host material can be removed reversibly to generate a stable empty host material. [15].
Porous materials have attracted the attention of chemists, physicists and material scientists not only for their industrial applications (separation, heterogeneous catalysis, and gas storage), but also because of the possibility of generating molecular assemblies which could be used for confining molecules [90]. Specific surface area is an important parameter for porous solids for which adsorbent and adsorbate interactions occur via a physisorption process. Two models Langmuir [92] and Brunauer-Emmett-Teller (BET) [93] are used to understand and measure the surface area of microporous materials from N2 or Ar gas adsorption isotherms. The Langmuir method is based upon monolayer coverage whilst the BET method is based upon multilayer coverage. As the adsorption mechanisms of these two models are different the values calculated from each method often differ for a specific material.
The adsorption of guest molecules onto a solid surface is fundamental in determining the properties of a porous compound [86]. This adsorption is governed not only by the interaction between guest molecules (adsorbates) and the surfaces (adsorbents), but also by the pore size and shape. Pores are classified according to their size (Table 1) [94].

Adsorption by a macropore and adsorption onto a single surface are similar [15, 85]. In a mesopore this phenomenon is dominated by capillary condensation which results in a sharp adsorption increase around the mid relative-pressure region [95] whereas in a microporous material should not be considered as molecules attaching onto a solid surface, but as the filling of molecules into a nanospace where a deep potential field is generated by the overlapping of the wall potentials [15]. In this case the adsorption isotherm shows a steep rise at very low relative pressure and a plateau after saturation. There are six distinct representative adsorption isotherms that reflect the relationship between porous structure and sorption type [96, 97]. The IUPAC classification of adsorption types is shown in Fig. 12. [96, 97]
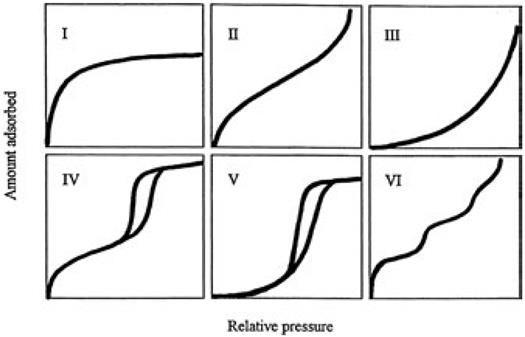
These adsorption isotherms are characteristic of adsorbents that are microporous (type I), nonporous and macroporous (types II, III and VI), and mesoporous (types IV and V). The differences between types II and III and between types IV and V arise from the relative strength of fluid-solid and fluid-fluid attractive interactions. When the fluid-solid attractive interactions are stronger than the fluid-fluid interactions, the adsorption isotherm should be type II or IV. The opposite situation leads to type III, and V isotherms. A type VI isotherm represents adsorption onto nonporous or macroporous solid surfaces where stepwise multilayer adsorption occurs.
Porous Coordination Polymers
Porous materials allow the reversible passage of host molecules through the guest materials via surface holes [97]. Some inorganic materials are commonly used such as zeolites and activated carbons. We target in this work coordination polymers that are both highly crystalline and porous, but it is well-known that these two properties are conflicting [16]. When cavities appear in these frameworks they are often filled by either solvent molecules, counter ions or additional co-crystallising organic molecules. These, molecules may act as templates which determine the pore shapes and sizes of the framework [42]. However, the bonds within the coordination polymer are non-covalent and so the removal of the template molecules may lead to the collapse of the structure [14].
If, after removal of the template molecules from a coordination polymer (desolvated) it remains crystalline and stable, only then can the material be classified as a porous coordination polymer [15]. To achieve permanent porosity we must initially target either a rigid or a flexible framework. A rigid framework [13] is where essentially no changes are observed between the full and empty host structures and a flexible framework [13] is where substantial movement of the host structure may occur with desolvation but without the loss of porosity.
Gas Storage in Porous Coordination Polymers
The safe storage and transport of gases in high concentration (at room temperature) requires compression at high pressures for gases such as CH4 and H2. This process consumes a great deal of energy and does not make for safe conveyance. As discussed previously, one solution to this problem is the use of solid adsorbents as carriers which can also perform at low gas pressures. Porous coordination polymers, which have a high percentage of uniform microporosity necessary for high efficiency in conjunction and a degree of tuneability, have emerged as candidates as solid adsorbents [86].
Gas storage potential is measured through sorption isotherms, but interest in developing single-crystal adsorbents has also led to structural studies of gas loaded framework materials [13]. These studies are also important in ascertaining the preferential binding sites for gas molecules within a framework [85]. When a gas molecule is transferred from the gas phase onto a solid surface (including pore surfaces) and remains on the surface, gas adsorption has occurred [97]. In contrast, gas absorption involving gas molecules taken into the solid phase is called occlusion [97].
There are two ways in which a gas can interact with a solid, chemisorption [97] in which gas molecules form chemical bonds with the solid, and physisorption [97] where the gas molecules interact with the surface of the solid without forming covalent bonds. The chemisorption process allows very high volume uptakes of gaseous molecules under ambient conditions, but requires energy input for gas release. Conversely, the weak interactions between gas and solid in the physisorption process often leads to lower volumes of gas uptake and occur only at low temperatures but are highly reversible kinetically and thermodynamically. Hence, both processes have their disadvantages. To achieve optimum gas adsorption within solid adsorbents porous materials must show intermediate strength interactions, termed pseudo chemisorption [85, 86].
The degree of interaction between a porous host and a gas molecule can be quantified by determination of the heat of adsorption (enthalpy of adsorption, ΔHads) [99]. This value is usually calculated by collecting adsorption data at least two temperatures, such as 78 and 88 K, and applying the Clausius-Clapeyron equation. Materials which exhibit strong chemisorption interactions, like metal hydrides, have high ΔHads values whereas materials which have weaker physisorption interactions, for example activated carbons and zeolites, show low ΔHads values [13]. Generally, when the, ΔHads is higher than 50 kJ mol-1[95], chemical adsorption is occurring. We target materials that store gas molecules at intermediate ΔHads values so that they adsorb and release the gas molecules at operational temperatures and pressure.
Porous coordination polymers have been found to be good candidates for the storage and separation of a range of gases [100, 101, 102] as they show appropriate ΔHads values and volumetric uptake. In comparison with other porous materials, the internal surfaces of porous coordination polymers are rich in hydrocarbons and aromatic groups which are known to attract guest molecules [85]. The tuneability of pore character allows optimum properties to be readily targeted. This is particularly important as the gases of interest (H2, CH4, CO2, C2H4, O2, etc.) differ in size, shape and chemical nature. Thus, the pore size, shape and character of the solid adsorbent must also be varied accordingly.
Methane Gas Storage in Porous Coordination Polymers
Methane (CH4) is the main component of natural gas and a candidate as a clean fuel for transportation [102]. The storage of CH4 on adsorbents is of interest as an alternative to high-pressure compressed gas storage. However, none of the conventional adsorbents have afforded sufficient CH4 storage to be commercially viable [16]. Activated carbons with a high specific surface area and containing micropores are not effective for CH4 adsorption due to a high percentage of mesopores and macropores also present in the structure [103]. This is because the surface in the meso/macro pores cannot trap CH4 molecules and therefore the presence of large voids does not enable gas storage. In addition, the energy density of adsorbed gas must be comparable to that of compressed natural gas used currently [103]. To achieve a higher adsorption capacity it is necessary to ensure that micropores of appropriate size and chemically compatible to adsorb CH4 molecules, are densely and uniformly distributed within the solid.
CH4 gas adsorption for porous coordination polymers was first reported by Kondo and co-workers [104] for {[Co2(4,4'bpy)3(NO3)4]·4H2O}∞, (bpy = 4,4'-bipyridine). Many early examples of methane storage in porous coordination polymers showed enhanced capacity compared with conventional materials such as zeolite 5A and activated carbon AX-21 [95]. Currently, many examples of frameworks [15] have been reported with ever greater storage capacities as a consequence of incorporating pores of an ideal size to maximise CH4 framework interactions.
Carbon Dioxide Gas Storage in Porous Coordination Polymers
Removal of carbon dioxide (CO2) from the exhaust of power plants is commonly accomplished by chilling and pressurising the waste gas or by passing the fumes through a fluidised bed of aqueous amine solution. Both approaches are costly and inefficient [105]. Alternatively, the chemisorption of CO2 on oxide surfaces or adsorption within porous silicates, carbons, or membranes have been considered but these technologies show no long-term viability [106]. In order to obtain an effective material with feasibility for CO2 removal it should combine two characteristics [107]. First, a periodic structure for which CO2 uptake and release is fully reversible, and second, the system must be flexible, chemically functionalised and fine-tuned at a molecular level such that optimised uptake capacities can be achieved.
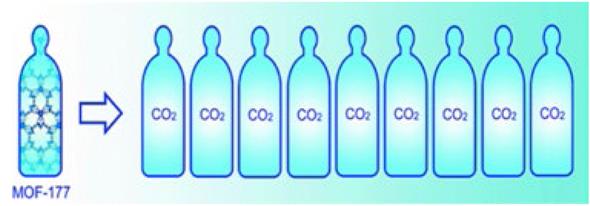
Porous coordination polymers have been shown to be promising candidates for CO2 storage already [108]. Yaghi et al. [107, 109] reported the porous coordination polymer {[Zn4O(BTB)2].(DEF)15(H2O)3}∞, (BTB = 4,4'',4'''-benzene1,3,5-triyl-tri-benzoate, MOF-177) that shows a high capacity of 150 wt %. This uptake represents a ninefold increase in CO2 uptake where MOF-177 [109] is present compared with when there is no solid adsorbent [107] (Fig. 13).
Acetylene Gas Storage in Porous Coordination Polymers
Acetylene (C2H2) is a key starting chemical for many chemical products [110] and materials [111] in the petrochemical and electronics industries. To obtain high purity C2H2 for the preparation of these materials it is important to separate C2H2 from the crude gas mixture, which also contains CO2, with a minimal cost in energy [112]. Zeolites and activated carbons are known to be capable of accommodating large amounts of both gases [103]. No significant discrimination between C2H2 and CO2 has been observed, because these molecules are similar to each other in equilibrium sorption parameters, and have related physico-chemical properties, molecular size and shape. C2H2 is known to be a highly reactive molecule and thus, it cannot be compressed above 0.2 MPa without risk of explosion even at room temperature [113].
Porous coordination polymers can be designed to adsorb specific molecules. For example, porous compounds with a cross-section of 4 to 6 Å are relevant for the accommodation of both C2H2 and CO2 molecules. Both molecules can be strongly confined in the pore without any chemical interaction, in other words, van der Waals potential energy [114]. Matsuda and co-workers [115] synthesised the complex {[Cu2(pzdc)2(pyz)]}∞, (pzdc = pyrazine-2,3-dicarboxylate and pyz = pyrazine) which shows high levels of selective sorption of C2H2 as compared to CO2. The enthalpy of adsorption values for C2H2 show a better interaction with the porous coordination polymer surface than for CO2 and thus, {[Cu2 (pzdc)2(pyz)]}∞, is well suited to perform gas separation.
Oxygen Gas Storage in Porous Coordination Polymers
Oxygen (O2) separation from air is an important industrial process [13]. The techniques used to perform this include three major types [14], cryogenic distillation, air separation at ambient temperatures and high-temperature air separation [116]. The adsorbent medium is often the major cost of adsorption separation technology, and thus research on improved adsorbent materials is of great interest. Porous coordination polymers are capable of increasing selectivity, improving energy efficiency and reducing the costs of the O2 separation. Kitagawa et al. [117] reported O2 physisorbed in the nanochannels of framework {[Cu2(pzdc)2(pyz)]}∞[117] (Fig. 14). In addition, the storage of O2 molecules in porous coordination polymers is of interest as the magnetic and redox properties of O2 may be altered due to the confined space. [117]

H2 Gas Storage in Porous Coordination Polymers
In 2006 the US Department of Energy (DOE) set a short-term could be used in automobile fuelling by 2010, and a long-term goal of 9.0 wt % and 81g L-1 by 2015 [118]. Another target was to store H2 at near ambient temperatures and pressures of <100 bar. From a practical perspective any potential materials must also be of minimal weight and volume for incorporation into vehicles. Additionally, H2 adsorption and desorption should be totally reversible and the refuelling of H2 should be completed within minutes [118]. Therefore, the volume weight percentage uptake is just as important as the kinetics and the thermodynamics of H2 release and uptake (Fig. 15).

Fig. 15 Heat of adsorption values observed for materials which undergo physisorption and those which undergo chemisorption. The target range for useful H2 storage is between 10 and 50 kJ mol-1.
Materials such as metal hydrides, which undergo chemisorption of H2 gas, generally show ΔHads > 50 kJ mol-1, which is too high for efficient gas release [85]. Materials such as carbon nanotubes, zeolites and activated carbons, show physisorption of H2 gas, ΔHads < 10 kJ mol-1, and this H2 adsorption can only occur at very low temperatures [103].
Porous coordination polymers show physisorption of H2 molecules. Due to the exceptional high porosity, uniform pore size, and intermediate ΔHads values, these materials have become good candidates for H2 storage. The H2 storage capability for porous coordination polymers was first reported by Rosi and co-workers [82] for the framework {[Zn4O(BDC)3]}∞, (MOF-5). These initial findings showed a low weight percentage uptake, 4.5 wt % at 77 K (pressure < 1 atm) and 1.0 wt % at room temperature and 20 bar [82, 119]. Since this first report of H2 adsorption on a MOF, over 200 porous coordination polymers have been investigated for H2 storage and they commonly outperform other porous materials [120, 121, 122]. Various strategies to enhance H2 uptake, such as the introduction of open metal sites, in porous coordination polymers at 78 K and 1 atm have been undertaken [121, 125]. The DOE targets for H2 storage describe systems operating at near-ambient temperatures and relatively high pressures (from 12 to 60 bar) [120, 126]. Thus, the current research for H2 storage in MOFs is directed towards high pressure systems (from 10 to 100 bar) [15, 85, 103]. In high pressure studies two concepts are used to describe H2 adsorption in porous coordination polymers the excess and total adsorption [85, 86]. Excess adsorption is the amount of adsorbed gas interacting with the framework and total adsorption is the amount of gas interacting with both the framework and any residual gas found in the pores of the porous coordination polymer in the absence of any gas-solid interaction [127].
Thus, the main goal is to generate porous coordination polymers with high H2 uptake and optimal ΔH ads values. The main characteristics reported thus far are a large pore volume/surface area for higher loadings and multiple sites for H2 adsorption [121-123, 128, 129].
Methods for Increasing the H2 Adsorption and the Heat of Adsorption
The design of porous frameworks towards high void volumes to effectively H2 store has be found in examples including the IRMOF series [130], which are some of the most porous materials reported in the literature, and the MIL series [131] which are giant pore systems (Fig. 16) [131, 132].
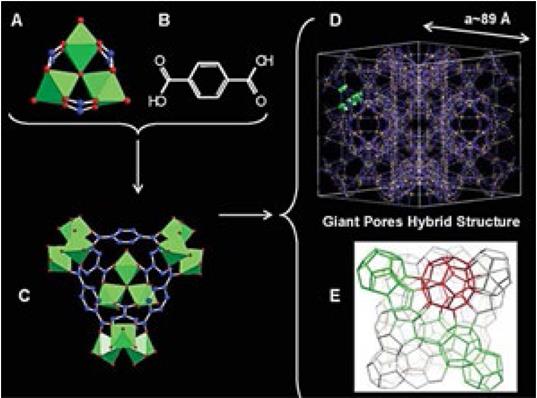
Fig. 16: (a) The trimeric building block chelated by three carboxylic functions; (b) terephthalic acid; (c) super tetrahedron; (d) ball-andstick representation of one unit cell; (e) schematic three-dimensional representation of the zeotype (From Férey, G.; et al. Science 2005, 309, 2040-2042.
Schröder et al. reported [61] a metal-organic framework, NOTT-112, that exhibits high total H2 storage (10 wt% at 77 bar and 77 K). The pore size distribution for NOTT-112 revealed a distribution of pores with estimated diameters of 7.2, 8.6, 10.9, 12.6 and 21 Å (Fig. 17). The BET surface area and the pore volumes were estimated to be 3800 m2 g-1 and 1.6 cm3 g-1 respectively. Thus, this combination of high surface and pore volume is necessary to achieve this level of H2 storage capacity.
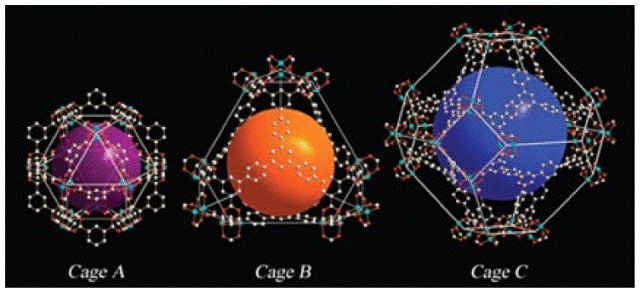
Fig. 17 Different cages in the crystal structure of NOTT-112. Copper: blue-green; carbon: grey; oxygen: red
Many porous coordination polymers adsorb H2 and a correlation between pore volume and H2 uptake capacity is observed [121, 133]. In particular, a study on a porous coordination polymers series [128] showed that H2 saturation uptake correlates with surface area. Thus, the maximum H2 storage capacity at 77 K depends linearly on the specific surface area of the framework and it is independent of composition and structure [128]. Although large pores are clearly effective to store large volumes of H2, small pores of 4.5 to 5 Å have been shown to allow a more efficient interaction of H2 with the surface of the pores. This increased interaction is important to permit the release of H2 at ambient temperatures [130]. A convenient way of generating small pores is through interpenetration of networks [84]. Kesanli and et al. [134] synthesised the framework {[Zn4(μ4-O)(L1)6](DMF)2}∞, (L1 =6,6'-dichloro-2,2'diethoxy-1,1'-binaphthyl-4,4'-dibenzoate) (Fig. 18). This system shows a fourfold interpenetrated porous coordination polymer with small pores and a surface area of 502 m2 g-1. Even though this material shows H2 adsorption of only 1.12 wt % at room temperature and 48 bar, the surface area is performing more efficiently in comparison to materials with a larger surface area. In addition, it is important to recognise that the interactions between the aromatic rings of the framework and H2 molecules lead to a greater H2 uptake.
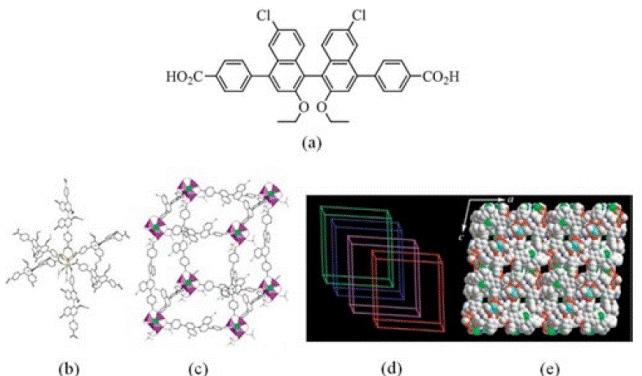
Fig. 18 (a) Schematic representation of the ligand 6,6'-dichloro-2,2'-diethoxy-1,1'-binaphthyl-4,4'-dibenzoic acid = H2L1; (b) view of the one of the {[Zn4(μ4-O)(L1)6](DMF)2}∞ clusters; (c) view of the cubic cavity formed by the 3D network; (d) schematic presentation of the fourfold interpenetration in {[Zn4(μ4-O)(L1)6](DMF)2}∞; (e) space-filling model of {[Zn4(μ4-O)(L1)6](DMF)2}∞ along the b axis (Kesanli, B., et al (2005), Highly Interpenetrated Metal-Organic Frameworks for Hydrogen Storage. Angew. Chem. Int. Ed., 44 (1).
The hypothesis that larger areas of aromatic surfaces led to increased H2 uptake was also investigated by Roswell and co-workers [129] in a study of five members of the IRMOFs series. In this work, the internal surface area and number of aromatic rings in the organic linker were varied systematically and a distinct correlation between H2 uptake and aromaticity was recorded.
Thus, there must be a compromise between the surface area and high crystal density when seeking porous coordination polymers with both high gravimetric and volumetric H2 uptakes [120, 129]. In addition, it is important that significant amounts of H2 are adsorbed at ambient temperatures so these materials can be used in industrial applications [85, 86, 135]. A binding energy of approximately 20 kJ mol-1 in the overall H2 loading range has been proposed as the optimum value for room temperature H2 storage [99, 135, 136].
Attempts to increase H2 adsorption enthalpies in PCPs, with the introduction of coordinatively unsaturated metal centres, open metal sites have been undertaken [135, 137, 138]. Initials reports of H2 storage in porous framework materials suggested that it was only aromatic surfaces towards which H2 molecules have an affinity [85]. However, further structural studies of the loading of H2 into porous coordination polymers showed that the preferred loading site, at very low pressure, is often an open metal site [139-143]. In addition, H2 binding to metal ions should lead to more efficient packing of the H2 molecules in the material, thus resulting in increased storage capacities [86]. This presents a scientific challenge as new synthetic methodologies must be devised to produce such systems.
One of the projects of the DOE's Hydrogen Storage sub-program works on the synthesis of new PCPs capable of achieving ~ 20 kJ mol-1, adsorption enthalpy, which required for their use as H2 storage materials operating under 100 bar at ambient temperatures. Their research includes both studying conventional PCPs as well as the synthesis of frameworks with the ability to bind additional metal centres. In the last Progress Report for the DOE Hydrogen and Fuel Cells Program [144], J. Long and M. Head-Godon reported the highest reliably-obtained isosteric heat of adsorption for a PCP: 13.7 kJmol-1 for Ni(m-dobdc). This framework surpassed and set the volumetric hydrogen storage record, with a capacity of 12.5 g L-1 at 298 K and 100 bar. Their study proved that the improvement in the H2 binding enthalpy is due to a change in the electronic structure at the open metal coordination sites.
Long and co-workers attempted to synthesise PCPs with the ability to bind additional metal centres and consequently bind, strongly, H2 and with a higher density. Thus, they synthesised one framework that can bin additional metal centres: the UIO-67-bipyridine dicarboxylate (bpydc) framework [144], which has a bipyridine in the linker. This PCP has been metalated with a variety of metal salts and several metalated samples showed higher H2 uptake than the bare framework at selected pressures. Based on the results of the adsorption experiments at 1 bar and 77 K, they estimate that there are 2.6 H2 molecules per metal centre for the sample PdCl2 and 1.5 for the CuCl2 sample. These findings set a potential method to enhance H2 uptake in PCPs and they have concluded that the most plausible method for achieving this it is post-synthetic incorporation of metal centres into ligands and use of these centres to interact with multiple H2 molecules. The new challenges are to develop ligands with charge-balancing functionalities for optimal metal binding properties and to achieve the binding of multiple H2 molecules per metal centre at room temperature.
A recently reported method for H2 adsorption enhancement is hydrogen spillover [145-147]. This is defined as the dissociative chemisorption of H2 onto a metal and the subsequent migration or surface diffusion of the atomic hydrogen onto a support surface (such as alumina) [148, 149]. This reaction is irreversible, with hydrogen atoms spontaneously recombining to afford the molecular gas (H2) [150]. Li and co-workers [145] showed that mechanically mixing 5% Pt on activated carbon with a MOF afforded materials with increased H2 adsorption and H2 ΔH ads.
Final Remarks on Gas Storage in Porous Coordination Polymers
Significant progress has been made since gas storage was first reported in a metal-organic framework [82]. In the last few years, considerable increases in the H2, CH4, C2H2 and CO2 binding energy and the storage capacity of porous coordination polymers have been reported [85, 135, 151-153]. Specific features such as high surface areas, the formation of narrow pores in the framework and free metal coordination sites on metal ions have been shown to enhance gas uptake in porous coordination polymers [61, 135, 151, 152, 154, 155, 156, 157]. However, further technological advances will be required in order to use these materials as on-board and stationary storage systems. Thus, the creation of porous coordination polymers for gas storage applications presents a formidable and complex challenge.

